一种双子酯基季铵盐表面活性剂及其合成方法与流程
本发明涉及一种双子酯基季铵盐表面活性剂及其合成方法。
背景技术:
双子表面活性剂是用联结基团,将两个普通表面活性剂分子的亲水头基或在靠近头基处以化学键方式联接在一起形成的一种新型表面活性剂。与传统单链表面活性剂相比,双子表面活性剂具有更为优异的表面活性和生物活性,成为化学界研究的重点和热点。
虽然传统单链、双子季铵盐表面活性剂化学性质稳定,具有抗菌、抗静电等良好性能,但其毒性大、难易降解,对生态环境造成较大的压力。一般来说,在表面活性剂分子的疏水链和亲水头基间引入酯基、酰胺基等可降解基团能有效提高其生物降解性。双子酯基季铵盐表面活性剂不仅具由传统双子表面的高表面活性,还因引入了酯基而具有高生物降解性能和低毒性,获得“绿色表面活性剂”的美誉。因此,国内外研究学者更加注重可降解的表面活性剂的研究。
中国在双子酯基季铵盐合成研究方面较晚,研究深入程度不及西方发达国家。目前,国内文献报道主要由酸与醇进行酯化反应引入酯基,而该方法需要甲苯、二甲苯等带水剂以促进反应向右进行,提高转化率,而苯、甲苯等为有毒、易挥发溶液;同时,该方法所需温度较高、时间较长、后续提纯复杂。
技术实现要素:
本发明的目的在于克服现有方法中合成双子酯基季铵盐表面活性剂存在的技术问题,提供一种双子酯基季铵盐表面活性剂及其合成方法。
本发明所采取的技术方案是:
一种双子酯基季铵盐表面活性剂,其化学结构式如式(ⅰ)所示:
式(ⅰ)中,n=13、15、17;m=2~6;x为f、cl、br或i。
优选的,式(ⅰ)中,n=15;m=4;x=br,即优选的一种双子酯基季铵盐表面活性剂的化学结构式为:
一种双子酯基季铵盐表面活性剂的合成方法,包括以下步骤:
1)将天然脂肪酸和卤化试剂进行酰化反应,得到脂肪酸酰卤;
2)将脂肪酸酰卤与n,n-二甲基乙醇胺(dmea)进行醇解反应,得到脂肪酸二甲氨基乙醇酯;
3)将脂肪酸二甲氨基乙醇酯与二卤代烷进行季铵化反应,得到式(ⅰ)所示结构的双子酯基季铵盐表面活性剂。
优选的,合成方法的步骤1)中,天然脂肪酸与卤化试剂的摩尔比为1:(1~1.5)。
优选的,合成方法的步骤1)中,酰化反应的温度为80℃~95℃,反应的时间为2h~5h;进一步优选的,酰化反应的温度为88℃~92℃,反应的时间为2.5h~3.5h。
优选的,合成方法的步骤1)中,酰化反应中加入催化剂参与反应;进一步的,酰化反应的催化剂为n,n-二甲基甲酰胺。
优选的,合成方法的步骤1)中,酰化反应是在回流条件下进行反应。
优选的,合成方法的步骤1)中,酰化反应后得到粗脂肪酸酰卤,再经旋转蒸发去除卤化试剂,得到除杂后的脂肪酸酰卤。
优选的,合成方法的步骤1)中,天然脂肪酸为c14~c18的脂肪酸;进一步优选的,天然脂肪酸为棕榈酸。
优选的,合成方法的步骤1)中,卤化试剂为氯化亚砜、磺酰氯、五氯化磷、三氯化磷、氯硅烷中的至少一种;进一步优选的,卤化试剂为氯化亚砜。
优选的,合成方法的步骤2)中,脂肪酸酰卤与n,n-二甲基乙醇胺的摩尔比为1:(1~2)。
优选的,合成方法的步骤2)中,醇解反应为在室温下反应4h~10h。
优选的,合成方法的步骤2)中,醇解反应中加入缚酸剂参与反应;进一步的,缚酸剂为三乙胺。
优选的,合成方法的步骤2)的醇解反应中,缚酸剂与脂肪酸酰卤的摩尔比为(1~1.5):1。
优选的,合成方法的步骤2)中,醇解反应在卤代烷溶剂中进行;进一步优选的,卤代烷为二氯甲烷、三氯甲烷、四氯化碳、1,2-二氯乙烷中的至少一种;再进一步优选的,卤代烷为二氯甲烷。
优选的,合成方法的步骤2)中,醇解反应后,减压过滤,并用卤代烷洗涤滤渣,合并滤液,将滤液减压蒸去溶剂、缚酸剂及原料,得到提纯后的脂肪酸二甲氨基乙醇酯。
优选的,合成方法的步骤3)中,脂肪酸二甲氨基乙醇酯与二卤代烷的摩尔比为1:(2~2.2)。
优选的,合成方法的步骤3)中,季铵化反应的温度为90℃~110℃,反应的时间为5h~10h;进一步优选的,季铵化反应的温度为98℃~102℃,反应的时间为5.5h~6.5h。
优选的,合成方法的步骤3)中,二卤代烷为二溴乙烷、1,3-二溴丙烷、1,4-二溴丁烷、1,5-二溴戊烷、1,6-二溴己烷中的任意一种;进一步优选的,二卤代烷为1,4-二溴丁烷。
优选的,合成方法的步骤3)中,季铵化反应在回流条件下进行反应。
优选的,合成方法的步骤3)中,季铵化反应是在低沸点溶剂中进行反应;进一步优选的,低沸点溶剂为甲醇、乙醇、丙醇、异丙醇、乙酸乙酯中的至少一种;再进一步优选的,低沸点溶剂为异丙醇。
优选的,合成方法的步骤3)中,季铵化反应后,经减压蒸馏溶剂得到粗产品,再通过重结晶的方法提纯得到双子酯基季铵盐表面活性剂产物;进一步优选的,重结晶试剂为异丙醇和乙酸乙酯混合溶剂。
本发明的有益效果是:
本发明采用价廉质高的原料经酰化、醇解、季铵化三步法合成可生物降解的双子酯基季铵盐表面活性剂,该方法成本低廉、反应条件温和、产率较高,所得的产物性能良好。本发明对双子酯基季铵盐表面活性剂的进一步开发、应用提供了基础数据支持。
与现有技术相比,本发明具有以下的优点:
1、使用的反应原料具有来源广、成本低廉、绿色安全、且可降解,对环境的危害小的特点。
2、合成过程中,在基团中引入酯键,赋予了产物优良的生物降解性能。
3、与将脂肪酸与醇直接反应相比,本合成方法反应条件温和、反应时间短、提纯步骤简单的优点。
附图说明
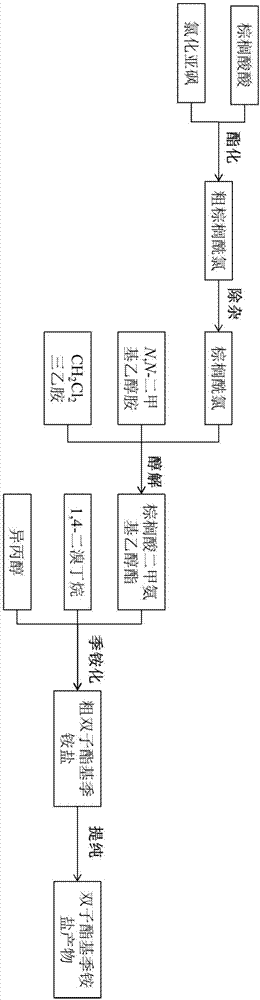
图1是实施例合成方法的流程示意图;
图2是实施例原料dmea、中间体和产物的红外光谱图;
图3是实施例产物的核磁共振氢谱图;
图4是实施例产物的热重图;
图5是实施例产物对苯的增溶性能图;
图6是市售产品十六烷基三甲基溴化铵对苯的增溶性能图。
具体实施方式
以下通过具体的实施例对本发明的内容作进一步详细的说明。实施例中所用的原料如无特殊说明,均可从常规商业途径得到。
实施例
附图1是本实施例合成方法的示意图,该图仅代表本发明合成方法的一个具体实施方式,本发明的合成原料不仅限于图中所示的物质。以下结合图1,对本实施例的方法作进一步具体的说明。
本实施例一种双子酯基季铵盐表面活性剂的合成方法,包括以下步骤:
1)制备棕榈酰氯:将棕榈酸和氯化亚砜进行酰化反应,得到棕榈酸酰氯
2)制备棕榈酸二甲氨基乙醇酯:将棕榈酸酰氯与n,n-二甲基乙醇胺进行醇解反应,得到棕榈酸二甲氨基乙醇酯
3)制备双子酯基季铵盐表面活性剂:将棕榈酸二甲氨基乙醇酯与1,4-二溴丁烷进行季铵化反应,得到双子酯基季铵盐表面活性剂丁撑基双(二甲基正十六酸乙酯基)溴化铵
具体反应步骤如下:
第一步酰化反应(棕榈酰氯的制备):向带有回流装置、磁力搅拌子和恒压滴液漏斗的250ml三口烧瓶中注入0.1mol棕榈酸,70℃保温至棕榈酸完全熔化后,加0.05ml催化剂dmf,再逐滴滴加15mlsocl2。滴加完毕后,升温至90℃,回流搅拌反应3小时,得到橘黄色的棕榈酰氯粗产品。将粗产品转至单口瓶后,旋转蒸发半小时除去多余的socl2,即可得较为纯净的棕榈酰氯,反应式如下:
第二步醇解反应(棕榈酸二甲氨基乙醇的制备):将0.1mol棕榈酰氯溶于40ml二氯甲烷后,于冰浴条件下,缓慢滴加于装有15mldmea和10.14g三乙胺的250ml三口烧瓶中,边滴加边剧烈搅拌,滴加完毕后,室温反应5小时。反应结束后,减压过滤除去三乙胺的盐酸盐,并用二氯甲烷洗涤滤渣,合并滤液。将滤液移至单口瓶后,减压蒸去溶剂、多余的缚酸剂及原料,得淡黄色液体棕榈酸二甲氨基乙醇酯,反应式如下:
先酰化后醇解的方法制备中间产物,经提纯后,最终得到产品质量为27.9g。
第三步季铵化反应(双子酯基季铵盐的制备):在装有温度计、回流装置的三口烧瓶中依次加入棕榈酸二甲氨基乙醇酯、1,4-二溴丁烷、异丙醇,室温搅拌15分钟后,测取初始胺值。将体系温度升至100℃,继续回流搅拌6小时,期间每小时测一次胺值。反应结束后,减压蒸出溶剂得到粗产品。以异丙醇/乙酸乙酯混合试剂为重结晶试剂进行多次重结晶得淡黄色固态的酯基gemini季铵盐产物,反应式如下:
对比例1——直接酯化法
用电子天平准确称量0.1mol棕榈酸后,并将其转移到装有磁力搅拌子的四口烧瓶中,插入温度计,搭建好分馏装置后,设置油浴锅为70℃,加热搅拌直到棕榈酸完全融化,再加入一定量的对甲苯磺酸和次磷酸作为催化剂,通入n2保护,用恒压漏斗滴加0.18moln,n-二甲基乙醇胺,待n,n-二甲基乙醇胺滴加结束后,将体系的温度升至160℃,搅拌反应,期间,每小时测定酸值,反应7h后酸值的变化幅度不大时停止反应,转化率为78.4%,计算得到25.679g。
待反应体系冷却至室温后,加入适量的质量分数为50%的koh溶液进行除杂,在75℃下搅拌反应半小时,之后将得到液体转移至梨形分液漏斗,注入适量的ccl4进行萃取(静置一晚),取出下层液体并将其进行旋蒸处理以除去萃取剂,即可得到淡黄色液态产品棕榈酸乙酯基二甲基叔胺,称重,16.8g。
对比可见,直接酯化法所需原料dmea的量较多、反应温度较高,而合成得到中间体产率不高,同时,提纯方法较为复杂、耗时较长、且提纯过程中中间体酯铵损失较多。另外,直接酯化合成需要温度条件控制较好,否则,酯铵颜色加深,由原来的淡黄色转变成橙黄色。而本发明先酰化后酯化的方法合成酯铵,反应所需原料dmea的量较少且只需在室温反应,且所得到最终产物较直接酯化的要高。总的来说,后者所需原料较少、反应条件温和、反应产率高。
对比例2——文献酯铵合成法
参照现有文献中的合成方法(王中才.酯基gemini型季铵盐表面活性剂的合成、性能及应用研究[d].江南大学,2005),进行酯铵合成法对比试验。
在带有搅拌器、球形冷凝管及滴液漏斗的250ml四颈烧瓶中,加入40.0g(0.20mol)十二酸,于50℃搅拌滴加28.5g(0.24mol)二氯亚砜,反应2小时后减压蒸除过量的二氯亚砜,再于85℃向所得产物中滴加21.4g(0.24mol)dmea,继续搅拌反应4小时,冷却至室温后加入70ml质量分数为8%的na2co3水溶液,充分搅拌,静置分层。有机层依次经饱和食盐水洗、无水硫酸镁干燥后过滤得淡黄色透明液体。
与现有文献的酯铵合成法对比可见,本方法所需温度低(室温);在体系中添加适量的缚酸剂三乙胺,则无需后续的碱洗、静置、盐水洗、干燥等步骤。另外,本发明人在实验中发现,反应的中间体极易与水溶液产生乳化现象、不易分离,而本方法免去水洗步骤,大大地避免乳化现象的发生。
表征分析
附图2是实施例原料(dmea)、中间体(棕榈酸二甲氨基乙醇酯)和产物(酯基gemini季铵盐)的红外光谱图。由图2可知,对比原料dmea、中间体棕榈酸二甲氨基乙醇酯以及产品的红外光谱图,发现产品已成功引入酯基、季铵基团。其中,2921cm-1、2851cm-1处为c-h伸缩振动吸收峰,1746cm-1处有强烈c=o吸收峰,1468cm-1处为-ch2-的c-h变形振动,1162cm-1处有长链脂肪酸c-o伸展振动,1080cm-1处为-c-n-伸缩振动吸收峰,721cm-1处是-(ch2)n-的摇摆振动吸收峰。所有官能团均吻合,可初步判断为丁撑基双(二甲基正十六酸乙酯基)溴化铵。
附图3是本实施例产物的核磁共振氢谱图。1hnmr(500mhz,dmso),δ=2.5(溶剂氘代dmso),δ=4.45(s,4h,-ch2-coo-),δ=3.65(s,4h,-n+-ch2-),δ=3.38(d,12h,ch3-n+-),δ=3.32(s,4h,n+-ch2-ch2-),δ=2.29(s,4h,-coo-ch2-),δ=1.70(s,4h,n+-ch2-ch2-),δ=1.64(s,4h,-coo-ch2-ch2-),δ=1.52(s,4h,-ch2-ch3),δ=1.24~1.19(s,44h,-(ch2)11-),δ=0.85(s,6h,-ch2-ch3)。图3的谱图符合目标产物的化学结构。
附图4是本实施例产物的热重图。热重分析结果表明,本发明产物的失重过程可分为四个阶段。当温度区间为60~105℃时,样品开始缓慢失重,失重率为1.211%,这主要是样品表面的少量水分蒸发导致的;随着温度的升高,当温度达到200~350℃时,样品质量剧烈下降,失重率达88.575%,这是碳碳键、碳氮键以及酯基于该温度区间内断裂、分解从而使样品质量下降;随着温度的进一步升高,失重速率趋于稳定,样品失重放缓,当温度区间为350~500℃时,失重率为92.489%;当温度区间为500~700℃,样品质量进一步下降,失重率达100%。与文献报道的传统单链以及不含酯基的双子季铵盐表面活性剂(开始剧烈失重温度为160℃左右)相比,产物开始剧烈失重的温度较高,这说明了引入酯基不一定会使双子表面活性剂的热稳定性降低,该表面活性剂具有良好的热稳定。
附图5、6分别是本实施例产物(代号16-4-16)以及市售产品十六烷基三甲基溴化铵(ctab)对苯的增溶性能图。由图5和图6对比可知,合成产物的增溶极限为a=0.391ml,计算得出本发明产物的增溶能力为x=3910ml/mol;而常规市售单链季铵盐表面活性剂ctab的增溶极限、增溶能力分别为0.137ml、1370ml/mol。可见,本发明产物的增溶能力明显优于ctab,这是因为与单链表面活性剂相比,双子表面活性具有更低的临界胶束浓度,胶团数和胶团内聚集数增加,胶团体积也随之增大,增溶于胶束内核的非极性烃类的量增多,表现为增溶能力增大。
本发明采用天然油脂提取的棕榈酸、价廉质高的n,n-二甲基乙醇胺、1,4-二溴丁烷为原料经酰化、醇解、季铵三步法合成可生物降解的双子酯基季铵盐表面活性剂丁撑基双(二甲基正十六酸乙酯基)溴化铵,该方法反应条件温和、产率较高。
与现有技术相比,本发明具有以下的优点:
1、使用的反应原料棕榈酸是从天然油脂中提取的,具有来源广、成本低廉、绿色安全、且可降解,对环境的危害小的特点。
2、使用的反应原料n,n-二甲基乙醇胺为一种用途广泛的化工原料,具有价廉质高的特点。
3、合成过程中,在基团中引入酯键,赋予了产物优良的生物降解性能。
4、与将脂肪酸与醇直接反应相比,本合成方法反应条件温和、反应时间短、提纯步骤简单的优点。
- 还没有人留言评论。精彩留言会获得点赞!