一种亲水凝胶骨架型缓控释固体制剂检测前处理方法与流程
本发明涉及分析化学
技术领域:
,具体涉及一种亲水凝胶骨架型缓控释固体制剂检测前处理方法。
背景技术:
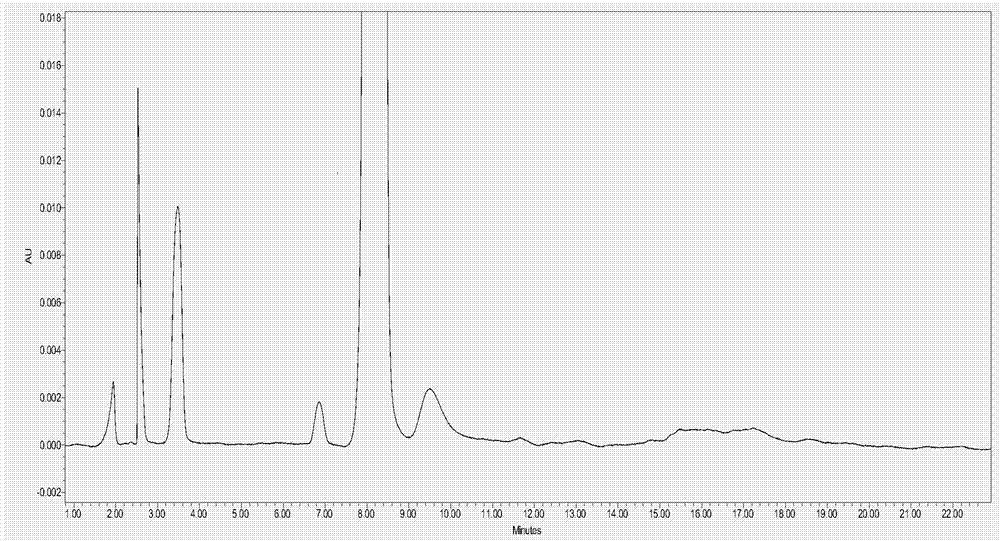
:口服缓控释制剂系指用药后能在较长时间内持续释放药物的制剂。缓控释制剂中的药物按适当的速度缓慢释放,血药浓度“峰谷”波动较小,可避免超过治疗血药浓度范围的毒副作用,又能较长时间保持在有效浓度范围之内以维持疗效。与普通制剂比较,缓控释制剂可延长治疗作用持续时间,降低毒副作用,减少用药次数,改善用药的依从性,因而越来越引起重视。亲水凝胶骨架缓控释口服固体制剂是口服缓控释固体制剂的一种,其特点是骨架材料遇水或胃肠液后膨胀,形成凝胶屏障而控制药物的释放,其机理包括控制药物通过凝胶层的扩散及凝胶的溶蚀。常用的骨架材料为不同规格的羟丙甲纤维素,其他如卡波姆、海藻酸钠、甲基纤维素、羟丙基纤维素、羧甲基纤维素钠等也有使用。然而由于亲水凝胶材料的应用,这些亲水凝胶遇水成凝胶状态,从而在药物检测的前处理过程中难以将其离心或过滤而溶清,使得针对于药物制剂中的原料药含量以及有关物质的检测变得比较困难。技术实现要素:有鉴于此,本发明的目的在于提供一种亲水凝胶骨架型缓控释固体制剂检测前处理方法,使得所述方法能够将亲水性凝胶骨架缓控释固体制剂制备形成流动性好和易溶清的溶液,且制得的溶液中不存在干扰含量和有关物质检测的成分,确保了含量和有关物质检测的专属性和准确性,并且在亲水凝胶骨架材料含量高达100%时,也能形成流动性较高和易溶清的溶液,从而达到分离检测的目的。为实现上述目的,本发明提供如下技术方案:一种亲水凝胶骨架型缓控释固体制剂检测前处理方法,包括:将亲水凝胶骨架型缓控释固体制剂粉碎后加入溶媒和酶溶液处理,然后离心或过滤,取上清或滤液用于检测;其中,所述酶为淀粉酶和/或纤维素酶,所述溶媒为磷酸缓冲液或有机溶剂的水溶液。本发明在亲水凝胶骨架型缓控释固体制剂检测前以适宜的溶媒配合淀粉酶和/或纤维素酶组成酶溶液进行处理,能够极大地提高固体制剂溶液的流动性,便于后续的检测操作。本发明所述溶媒根据固体制剂中活性药物的水溶性来选择是磷酸缓冲液(优选ph为3.0的磷酸缓冲液)还是有机溶剂的水溶液,水溶液较好的活性药物可选择磷酸缓冲液,水溶性较差的活性药物选择有机溶剂的水溶液。作为优选,本发明所述磷酸缓冲液ph值为3.0,所述有机溶剂为甲醇或乙腈;更优选地,所述有机溶剂的体积百分比浓度为20-100%,即可以是甲醇、乙腈或它们的水溶液。在本发明的一些具体实施方式中,所述有机溶剂的水溶液可选择为20%的乙腈水溶液、30%的乙腈水溶液或25%的甲醇水溶液。作为优选,所述酶溶液为酶的磷酸缓冲液,所述磷酸缓冲液ph值优选为3.0;所述酶溶液中酶的浓度为0.05-0.5u/ml,进一步优选为0.1-0.5u/ml,更优选为0.3-0.5u/ml。当所述酶为纤维素酶和淀粉酶时,两者各自的浓度均采用上述酶浓度的优选方案。在本发明的一些具体实施方式中,所述酶溶液中酶的浓度为0.5u/ml。本发明所针对的前处理对象是采用亲水凝胶骨架材料制备成的固体制剂,其中的亲水凝胶骨架材料包括但不仅限于羟丙甲纤维素、卡波姆、羟丙纤维素、海藻酸钠、壳聚糖、羧甲基纤维素钠、甲基纤维素;而固体制剂中的活性药物可以是任何适宜的化合物,如盐酸氨溴索、单硝酸异山梨酯、盐酸二甲双胍、盐酸坦索罗辛等。在前处理中,作为优选,所述溶媒和酶溶液的体积比为(1-4):1;所述酶溶液的用量为每1g亲水凝胶骨架型缓控释固体制剂加入1-200ml酶溶液,在本发明的一些具体实施方式中,亲水凝胶骨架型缓控释固体制剂可加入如下体积的酶溶液;1g亲水凝胶骨架型缓控释固体制剂加入10ml酶液;350mg亲水凝胶骨架型缓控释固体制剂加入5ml酶液;50mg亲水凝胶骨架型缓控释固体制剂加入10ml酶液;1.25g亲水凝胶骨架型缓控释固体制剂加入10ml酶液;所述处理优选为振荡处理1-24h,更优选为3-8h,在本发明具体实施方式中可选择为3、5或8h。本发明采用市售的盐酸普拉克索缓释片(亲水凝胶骨架材料羟丙甲纤维素)和单硝酸异山梨酯缓释片(亲水凝胶骨架材料羟丙甲纤维素和羧甲基纤维素钠)为试验对象,对照组为仅采用溶媒稀释,结果显示,对照组所获得待测溶液存在流动性差,有大量不溶物的现象,导致检测无法开展或检测困难,而本发明获得的待测溶液具有相当高的流动性且溶液易通过离心或过滤溶清,可以用在诸如hplc、气相色谱等检测中。此外,本发明还以100%的亲水凝胶骨架材料为试验对象,对照组为加溶媒稀释辅助振荡或超声溶解,结果显示,对照组普遍成凝胶状,无法过滤和离心,而具有流动性的对照过滤困难,无法获得澄清溶液。而本发明所处理的试验组溶液流动性极好,过滤和离心操作可顺利进行,所得待测溶液澄清。以市售的盐酸二甲双胍缓释片为测试对象,不同的处理方式检测其含量以及有关物质,结果显示只有经过本发明所述前处理方法处理后才能准确的检测。由以上技术方案可知,本发明通过适宜的溶媒和酶溶液对亲水凝胶骨架材料制备成的固体制剂进行处理,所获得溶液具有极佳的流动性和易溶清,溶清后的溶液中不存在干扰后续药物制剂中的原料药含量以及有关物质检测的成分,确保了制剂含量和有关物质检测的专属性和准确性。附图说明图1所示为市售单硝酸异山梨酯缓释片的hplc谱图;图2所示为图1的局部放大hplc谱图;图3所示为市售盐酸二甲双胍缓释片含量检测的hplc谱图;图4所示为市售盐酸二甲双胍缓释片原料药有关物质检测的hplc谱图;图5所示为图4的局部放大hplc谱图;图6所示为市售盐酸二甲双胍缓释片采用处理方式2所得样品有关物质检测的hplc谱图;图7所示为图6的局部放大hplc谱图;图8所示为市售盐酸二甲双胍缓释片采用处理方式3所得样品有关物质检测的hplc谱图;图9所示为图8的局部放大hplc谱图;图10所示为市售盐酸二甲双胍缓释片采用处理方式4所得样品有关物质检测的hplc谱图;图11所示为图10的局部放大hplc谱图。具体实施方式本发明公开了一种亲水凝胶骨架型缓控释固体制剂检测前处理方法,本领域技术人员可以借鉴本文内容,适当改进工艺参数实现。特别需要指出的是,所有类似的替换和改动对本领域技术人员来说是显而易见的,它们都被视为包括在本发明。本发明的方法已经通过较佳实施例进行了描述,相关人员明显能在不脱离本
发明内容、精神和范围内对本文所述的的化合物和制备方法进行改动或适当变更与组合,来实现和应用本发明技术。下面结合实施例,进一步阐述本发明。实施例1:对市售盐酸普拉克索缓释片样品前处理的对比1、市售盐酸普拉克索缓释片组分盐酸普拉克索、羟丙甲纤维素、预胶化淀粉、微晶纤维素、硬脂酸镁;2、不同前处理方法(1)相关试剂稀释剂1:纯化水:乙腈=80:20;稀释剂2:纯化水:乙腈=20:80;稀释剂3:纤维素酶溶液(0.1u/ml):称取纤维素酶适量,用磷酸盐缓冲液(ph3.0)溶解并稀释制成浓度为0.1u/ml的溶液;;常规技术1:称取适量样品(羟丙甲纤维素约1g)置100ml量瓶中,加50ml稀释剂1,超声30min,样品成凝胶状,无法离心和过滤。常规技术2:称取适量样品(羟丙甲纤维素约1g)置100ml量瓶中,加50ml稀释剂2,超声30min,样品流动性差,离心和过滤非常困难。本发明方法:称取适量样品(羟丙甲纤维素约1g)置100ml量瓶中,加40ml稀释剂1和10ml稀释剂3,振摇3小时,样品具有很好的流动性,离心和过滤很容易。3、结果常规技术1和2中,样品溶液流动性均非常差,过滤和离心困难,导致检测无法开展或检测困难。而在本发明方法中,样品具有很好的流动性,离心和过滤很容易,检测容易进行。实施例2:市售单硝酸异山梨酯缓释片的含量和有关物质检测1、市售单硝酸异山梨酯缓释片组成表12、供试品制备过程2.1药粉制备:取20片上述片剂于研钵中,研磨成细粉。2.2供试品制备:稀释剂1:纯化水:甲醇=75:25;稀释剂2:甲醇;稀释剂3:纤维素酶和淀粉酶的混和酶溶液(各0.3u/ml):称取纤维素酶和淀粉酶各适量,用磷酸盐缓冲液(ph3.0)溶解并稀释制成浓度各为0.3u/ml的溶液,即得;常规技术1:称取样品408mg(约含亲水凝胶骨架350mg,约含单硝酸异山梨酯30mg),置100ml量瓶中,加稀释剂1超声,样品成凝胶状,无法离心和过滤。常规技术2:称取样品408mg(约含亲水凝胶骨架350mg,约含单硝酸异山梨酯30mg),置100ml量瓶中,加稀释剂2超声,样品具较好的流动性,但存在大量不溶物,很难过滤,离心液很难得到澄清溶液。本发明方法:称取样品408mg(约含亲水凝胶骨架350mg,约含单硝酸异山梨酯30mg),置100ml量瓶中,加20ml稀释剂1和5ml稀释剂3,振摇5小时,再加稀释剂1至刻度,摇匀。样品具有很好的流动性,过滤很容易。3、含量和有关物质测定hplc条件流动相:甲醇:水(25:75)流速:1.0ml/min,波长:210nm,柱温:25℃,进样量10μl,色谱柱十八烷基硅烷键合硅胶。4、检测结果采用常规技术1处理时,样品成凝胶状,无法离心和过滤;采用常规技术1处理时,样品具较好的流动性,但存在大量不溶物,很难过滤,离心液很难得到澄清溶液,无法进行hplc检测;采用本发明方法处理时,样品具有很好的流动性,过滤很容易。测得最大单杂为0.14%,总杂为0.26%。采用本发明方法处理所得样品含量和有关物质检测色谱图见图1,图1局部放大色谱图见图2。实施例3:亲水凝胶骨架材料混合物在不同前处理方法中的流动性1、亲水凝胶骨架材料混合物组成表2成分用量(g)羟丙甲纤维素(k100m)3羧甲基纤维素钠3卡波姆3羟丙基纤维素3海藻酸钠3壳聚糖32、取上述混合物1g(即含亲水凝胶骨架1g),置100ml容量瓶中,按下述方式处理:常规技术1:加纯化水搅拌和超声,样品成凝胶状,无法离心和过滤。常规技术2:加20%乙腈超声,样品成凝胶状,无法离心和过滤。常规技术3:加70%乙腈超声,样品成凝胶状,无法离心和过滤。常规技术4:加70%乙腈振荡8h,样品成凝胶状,无法离心和过滤。常规技术5:加乙腈超声,样品具流动性,但过滤极困难,离心难以得到澄清溶液。本发明方法:加20%乙腈30ml和10ml纤维素酶和淀粉酶的混和酶溶液(参照实施例2中稀释剂3),振荡8h,样品具有很好的流动性,过滤很容易。3、检测结果采用常规技术1~4处理得到的样品均成凝胶状,无法离心和过滤;采用常规技术5处理得到的样品具流动性,但过滤极困难,离心难以得到澄清溶液;采用本申请方法处理得到的样品具有很好的流动性,过滤很容易。实施例4:市售盐酸二甲双胍缓释片含量检测1.市售盐酸二甲双胍缓释片组分盐酸二甲双胍、羟丙甲纤维素、乙基纤维素、玉米淀粉、硬脂酸;2.供试品制备过程2.1药粉制备:取20片上述片剂于研钵中,研磨成细粉。2.2供试品制备:稀释剂1:50%甲醇;稀释剂2:纤维素酶和淀粉酶的混和酶溶液(各0.5u/ml):称取纤维素酶和淀粉酶各适量,用磷酸盐缓冲液(ph3.0)溶解并稀释制成浓度各为0.5u/ml的溶液,即得;稀释剂3:纤维素酶和淀粉酶的混和酶溶液(各50u/ml):称取纤维素酶和淀粉酶各适量,用磷酸盐缓冲液(ph3.0)溶解并稀释制成浓度各为50u/ml的溶液,即得;处理方式1:称取称取样品60mg(约含亲水凝胶骨架50mg,约含盐酸二甲双胍10mg),置100ml量瓶中,加50%甲醇20ml,振荡5h,加50%甲醇至刻度,样品成凝胶状,无法离心和过滤。处理方式2(本发明处理方法):称取称取样品60mg(约含亲水凝胶骨架50mg,约含盐酸二甲双胍10mg),置100ml量瓶中,加10ml稀释剂2和10ml甲醇,振荡5h,加50%甲醇至刻度,样品具有很好的流动性,过滤很容易。处理方式3:称取称取样品60mg(约含亲水凝胶骨架50mg,约含盐酸二甲双胍10mg),置100ml量瓶中,加10ml稀释剂3和10ml甲醇,振荡5h,加50%甲醇至刻度,样品同样具有很好的流动性,过滤也很容易。处理方式4:称取称取样品60mg(约含亲水凝胶骨架50mg,约含盐酸二甲双胍10mg),置100ml量瓶中,加10ml稀释剂2和10ml甲醇,振荡30h,加50%甲醇至刻度,样品同样具有很好的流动性,过滤也很容易。3、含量测定hplc条件流动相:0.05%庚烷磺酸钠溶液(用磷酸调节ph值至3.0)-乙腈(80:20)流速:1.0ml/min,波长:233nm,柱温:35℃,进样量10μl,色谱柱十八烷基硅烷键合硅胶。4、检测结果处理方式2所测平行两个样的含量结果分别为100.06%、99.91%。处理方式3所测平行两个样的含量结果分别为88.36%、92.59%。处理方式4所测平行两个样的含量结果分别为115.31%、107.27%。采用处理方式3所测含量结果明显偏低,且两个平行样间含量结果差异较大。浓度过高的酶溶液(稀释剂3中酶溶液为50u/ml)可能对样品中活性成分存在吸附。而处理方式4所测含量结果偏高,过长的酶解时间可能产生了干扰活性成分的物质。为了验证过高的酶溶液是否对活性成分存在吸附以及过长的酶解时间是否产生了干扰活性成分的物质,采用了空白辅料中加入定量活性成分的方式考察了不同处理方式的加标回收率。所得结果如下:处理方式3回收率为90.7%,确证了过高的酶溶液对活性成分存在吸附现象。处理方式4回收率为109.1%,确证了过长的酶解时间产生了干扰活性成分的物质。采用处理方式2所得样品含量检测色谱图见图3。实施例5:市售盐酸二甲双胍缓释片有关物质检测1.市售盐酸二甲双胍缓释片组分:同实施例42.供试品制备过程2.1药粉制备:取20片上述片剂于研钵中,研磨成细粉。2.2供试品制备:稀释剂1:50%甲醇;稀释剂2:纤维素酶和淀粉酶的混和酶溶液(各0.5u/ml):称取纤维素酶和淀粉酶各适量,用磷酸盐缓冲液(ph3.0)溶解并稀释制成浓度各为0.5u/ml的溶液,即得;稀释剂3:纤维素酶和淀粉酶的混和酶溶液(各50u/ml):称取纤维素酶和淀粉酶各适量,用磷酸盐缓冲液(ph3.0)溶解并稀释制成浓度各为50u/ml的溶液,即得;处理方式1:称取称取样品1.25g(约含亲水凝胶骨架1g,约含盐酸二甲双胍250mg),置50ml量瓶中,加50ml稀释剂1,振荡5h,样品成凝胶状,无法离心和过滤。处理方式2(本发明处理方法):称取称取样品1.25g(约含亲水凝胶骨架1g,约含盐酸二甲双胍250mg),置50ml量瓶中,加10ml稀释剂2和10ml甲醇,振荡5h,加50%甲醇至刻度,样品具有很好的流动性,过滤很容易。处理方式3:称取称取样品1.25g(约含亲水凝胶骨架1g,约含盐酸二甲双胍250mg),置50ml量瓶中,加10ml稀释剂2和10ml甲醇,振荡30h,加50%甲醇至刻度,样品同样具有很好的流动性,过滤也很容易。处理方式4:称取称取样品1.25g(约含亲水凝胶骨架1g,约含盐酸二甲双胍250mg),置50ml量瓶中,加10ml稀释剂3和10ml甲醇,振荡5h,加50%甲醇至刻度,样品同样具有很好的流动性,过滤也很容易。同时,取盐酸二甲双胍原料药250mg置50ml量瓶中,加50%甲醇溶解并至刻度,作为控制样品。3、有关物质测定hplc条件流动相:1.7%磷酸二氢铵溶液(用磷酸调节ph值至3.0)流速:1.0ml/min,波长:218nm,柱温:30℃,进样量20μl,色谱柱磺酸基阳离子交换键合硅胶为填充剂的色谱柱。4、检测结果处理方式最大单杂总杂处理方式20.04%0.23%处理方式30.43%0.95%处理方式40.002%0.013%控制样品(原料药)0.04%0.21%处理方式3所得有关物质结果明显较处理方式2偏大,远远超出了质量标准中限度(《中国药典》2015版盐酸二甲双胍:单杂0.1%,总杂0.5%)。在处理方式3所得色谱图中,出现几个较明显的色谱峰。这些色谱峰可能由于振荡时间过长,酶自身降解或酶水解辅料产生。处理方式4所得有关物质结果明显较处理方式2偏小。参考实施例4,浓度过高的酶溶液除了对样品中活性成分存在吸附外,对杂质也存在吸附,导致杂质检测结果偏低。其中,原料药有关物质检测色谱图见图4,局部放大图见图5;采用处理方式2所得样品有关物质检测色谱图见图6,局部放大图见图7;采用处理方式3所得样品有关物质检测色谱图见图8,局部放大图见图9;采用处理方式4所得样品有关物质检测色谱图见图10,局部放大图见图11。以上所述仅是本发明的优选实施方式,应当指出,对于本
技术领域:
的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1