一种提取芳香胺的方法、检测方法、试剂盒及用途与流程
本发明属于检测分析领域,具体涉及一种提取芳香胺的方法,还涉及检测芳香胺的方法、试剂盒及其用途。
背景技术:
芳香胺化合物大多具有毒性,常作为中间体存在于合成染料、杀虫剂、橡胶、塑料、粘合剂和药物等产品中。其中,有些芳香胺的毒性很强,有些则是潜在的致癌物,比如4-氨基联苯和2-萘胺就是熟知的致癌物。抽吸烟草制品产生的烟气中以及烟草和烟草制品中都存在芳香胺,但烟草和烟草制品中的芳香胺含量较烟气中更低。因此,准确测定烟草和烟草制品中的芳香胺含量对产品的安全评价有一定意义。
迄今为止,研究人员已对抽吸烟草制品产生的烟气中的芳香胺化合物进行了检测,检测方法包括气相色谱法(GC)、液相色谱法(LC)、毛细管电泳法和气相色谱-质谱联用法(GC/MS)等。其中,气相色谱法、高效液相色谱法易受杂质干扰影响,产生假阳性问题;气相色谱-质谱联用法虽可避免出现假阳性问题,但对于含复杂成分的样品,需在样品前处理中进行多次萃取、衍生化和过柱净化,操作过程复杂、周期较长。
与抽吸烟草制品产生的烟气相比,烟草和烟草制品中含有更多的成分,背景更为复杂,检测分析时的杂质干扰更为严重,并且由于烟草和烟草制品中的芳香胺含量更低,因此,更加不容易准确测定芳香胺的含量。迄今还未发现能对烟草或烟草制品中的芳香胺进行准确测定的方法。
目前,尚需一种检测芳香胺的方法,以准确定量烟草或烟草制品中的芳香胺。
技术实现要素:
本发明人创造性地获得了一种提取芳香胺的方法,相比于现有的提取方法,该方法能更大程度地提取出烟草和/或烟草制品中的芳香胺化合物,提取率更高。同时,本发明人还得到了一种检测芳香胺的方法,能够对烟草和/或烟草制品中的芳香胺化合物进行快速、准确的定量分析,并且检出限低,灵敏度更高,重现性好,操作更为简便。
本发明第一方面涉及一种提取芳香胺的方法,包括如下步骤,
(1)用强酸溶液浸提样品,分离得到初提液;所述样品为烟草和/或烟草制品;
(2)将初提液的pH值调整至大于7,用脂肪醚进行萃取,分离得到萃取液。
本发明第一方面的一个实施方式中,还包括如下步骤,
(3)去除萃取液中的脂肪醚,得到萃取物,将所述萃取物溶解于甲醇,得到提取液。
在一个实施方案中,步骤(3)中,通过将萃取液进行干燥来去除脂肪醚;优选地,干燥方式为氮气吹干。
在一个实施方案中,步骤(3)中,甲醇的体积用量为萃取液体积的50%-200%,优选为80%-120%,更优选为100%。
在一个实施方案中,步骤(3)中,去除脂肪醚前,还包括向萃取液中加入弱酸或弱酸溶液进行混合的步骤。
在一个实施方案中,所述弱酸为醋酸;优选地,所述醋酸的纯度为99%-100%(W/W),更优选为99.5%(W/W)。
在一个实施方案中,所述弱酸溶液为醋酸水溶液;优选地,所述醋酸水溶液的浓度不超过99%(W/W),更优选为40%-70%(W/W)、50%(W/W)、60%(W/W)或80%(W/W)。
在一个实施方案中,所述弱酸溶液(以弱酸计)或弱酸的添加量为0.1-0.8mL/(mL萃取液),更优选为0.3-0.7mL/(mL萃取液),进一步优选为0.5mL/(mL萃取液)。
本发明第一方面的一个实施方式中,包括如下A至M中的任意一项或者多项:
A.步骤(1)中,所述强酸溶液为强酸水溶液;优选地,所述强酸水溶液选自盐酸水溶液和硝酸水溶液中的一种或多种;
B.步骤(1)中,所述强酸溶液为稀溶液;
C.步骤(1)中,所述强酸溶液的浓度为1%-10%(W/W),优选为2%-8%(W/W),更优选为5%(W/W);
D.步骤(1)中,所述浸提的料液比为1:(20-100)(g/mL),优选为1:(40-70)(g/mL),更优选为1:50(g/mL);
E.步骤(1)中,浸提时间为10-150分钟,优选为20-100分钟,更优选为30分钟;
F.步骤(1)中,所述浸提在超声条件下进行;
G.步骤(2)中,将所述初提液的pH值调整至大于8,优选为大于10,更优选为13以上;
H.步骤(2)中,通过向初提液中加入固体碱或碱溶液来调整pH值,优选加入固体碱;更优选地,固体碱选自氢氧化钠和氢氧化钾中的一种或多种;
I.步骤(2)中,萃取前,还包括将调整完pH值的初提液静置冷却的步骤;优选地,静置冷却的时间为5分钟;
J.步骤(2)中,所述脂肪醚为极性脂肪醚,优选为低沸点的极性脂肪醚,更优选沸点为40-70℃,进一步优选沸点为50-60℃;优选地,所述脂肪醚选自乙醚和叔丁基甲醚中的一种或多种,更优选为叔丁基甲醚;
K.步骤(2)中,所述脂肪醚的用量为5-40mL/(g样品),优选为10-30mL/(g样品),更优选为20mL/(g样品);
L.步骤(2)中,萃取时间为5-20分钟,优选为10分钟;
M.步骤(1)中,所述芳香胺选自邻氨基偶氮甲苯、4-氨基联苯、2-氨基-4-硝基甲苯、联苯胺、对氯苯胺、4-氯-邻甲基苯胺、2-甲氧基-5-甲基苯胺、2-奈胺、4,4'-二氨基二苯基甲烷、2,4-二氨基甲苯、3,3'-二氯联苯胺、3,3'-二甲氧基联苯胺、3,3'-甲基-4,4'-二氨基二苯甲烷、4,4'-亚甲基-二-(2-氯苯胺)、4,4'-二氨基二苯醚、邻甲苯胺、4-氨基偶氮苯、2-氨基苯甲醚、2,4-二甲基苯胺和2,6-二甲基苯胺中的一种或多种。
本发明第二方面涉及一种检测芳香胺的方法,包括按照本发明第一方面任一项所述的提取方法获得萃取液和/或提取液的步骤;还包括用液相色谱串联质谱法测定萃取液和/或提取液的步骤。
本发明第二方面的一个实施方式中,所述测定为定量检测,优选为内标法定量检测;更优选地,内标物为d7-1-氨基萘。
本发明第二方面的一个实施方式中,液相色谱的操作条件包括如下a至d中的任意一项或者多项:
a.色谱柱为菲罗门Agilent ZORBAX SB-C3液相色谱柱;优选地,色谱柱的规格为150mm×2.1mm i.d.,5μm粒径;
b.色谱柱的温度为25-50℃,优选为30-50℃,更优选为40℃;
c.进样量为1-10μL,优选为2-8μL,更优选为3μL;
d.流动相由流动相A和流动相B组成,流动相A为甲醇,流动相B为0.1%(V/V)乙酸水溶液;优选地,流动相的洗脱程序如下表所示,
本发明第二方面的一个实施方式中,质谱的操作条件或参数包括如下1)至66)中的任意一项或者多项:
1)离子源为电喷雾电离源;
2)扫描方式为正离子扫描;
3)检测方式为多反应监测;
4)电喷雾电压为4000-7000V,优选为5000-6000V,更优选为5500V;
5)离子源温度为500-750℃,优选为550-700℃,更优选为650℃;
6)辅助气Gas1和/或Gas2的压力为40-70psi,优选为50-60psi,更优选为50psi或60psi;
7)邻氨基偶氮甲苯的定量离子对为226.1/91.1;
8)邻氨基偶氮甲苯的定性离子对为226.1/121.1;
9)邻氨基偶氮甲苯的碰撞能为20V;
10)4-氨基联苯的定量离子对为170.1/153.2;
11)4-氨基联苯的定性离子对为170.1/152.1;
12)4-氨基联苯的碰撞能为20V;
13)2-氨基-4-硝基甲苯的定量离子对为153.1/107.1;
14)2-氨基-4-硝基甲苯的定性离子对为153.1/89.9;
15)2-氨基-4-硝基甲苯的碰撞能为25V;
16)联苯胺的定量离子对为184.1/156;
17)联苯胺的定性离子对为184.1/166;
18)联苯胺的碰撞能为40V;
19)对氯苯胺的定量离子对为128/93.1;
20)对氯苯胺的定性离子对为128/111.2;
21)对氯苯胺的碰撞能为20V;
22)4-氯-邻甲基苯胺的定量离子对为142/107.2;
23)4-氯-邻甲基苯胺的定性离子对为142/125.2;
24)4-氯-邻甲基苯胺的碰撞能为23V;
25)2-甲氧基-5-甲基苯胺的定量离子对为138.1/123.2;
26)2-甲氧基-5-甲基苯胺的定性离子对为138.1/105.2;
27)2-甲氧基-5-甲基苯胺的碰撞能为20V;
28)2-奈胺的定量离子对为144.1/127.1;
29)2-奈胺的定性离子对为144.1/77.2;
30)2-奈胺的碰撞能为25V;
31)4,4’-二氨基二苯基甲烷的定量离子对为199.1/106.2;
32)4,4’-二氨基二苯基甲烷的定性离子对为199.1/77.2;
33)4,4’-二氨基二苯基甲烷的碰撞能为30V;
34)2,4-二氨基甲苯的定量离子对为123.1/108;
35)2,4-二氨基甲苯的定性离子对为123.1/106.1;
36)2,4-二氨基甲苯的碰撞能为23V;
37)3,3’-二氯联苯胺的定量离子对为253.1/217.1;
38)3,3’-二氯联苯胺的定性离子对为253.1/182.1;
39)3,3’-二氯联苯胺的碰撞能为30V;
40)3,3’-二甲氧基联苯胺的定量离子对为245.2/230.2;
41)3,3’-二甲氧基联苯胺的定性离子对为245.2/213.2;
42)3,3’-二甲氧基联苯胺的碰撞能为25V;
43)3,3’-甲基-4,4’-二氨基二苯甲烷的定量离子对为227.2/120.2;
44)3,3’-甲基-4,4’-二氨基二苯甲烷的定性离子对为227.2/178.2;
45)3,3’-甲基-4,4’-二氨基二苯甲烷的碰撞能为25V;
46)4,4’-亚甲基-二-(2-氯苯胺)的定量离子对为267.2/231.2;
47)4,4’-亚甲基-二-(2-氯苯胺)的定性离子对为267.2/140.2;
48)4,4’-亚甲基-二-(2-氯苯胺)的碰撞能为30V;
49)4,4’-二氨基二苯醚的定量离子对为201.1/108.3;
50)4,4’-二氨基二苯醚的定性离子对为201.1/184;
51)4,4’-二氨基二苯醚的碰撞能为23V;
52)邻甲苯胺的定量离子对为108.1/91.4;
53)邻甲苯胺的定性离子对为108.1/93.2;
54)邻甲苯胺的碰撞能为23V;
55)4-氨基偶氮苯的定量离子对为198.2/77.2;
56)4-氨基偶氮苯的定性离子对为198.2/51.2;
57)4-氨基偶氮苯的碰撞能为23V;
58)2-氨基苯甲醚的定量离子对为124.4/109.2;
59)2-氨基苯甲醚的定性离子对为124.1/92.3;
60)2-氨基苯甲醚的碰撞能为23V;
61)2,4-二甲基苯胺的定量离子对为122.1/107.2;
62)2,4-二甲基苯胺的定性离子对为122.1/105.2;
63)2,4-二甲基苯胺的碰撞能为23V;
64)2,6-二甲基苯胺的定量离子对为122.1/105.3;
65)2,6-二甲基苯胺的定性离子对为122.1/107.2;
66)2,6-二甲基苯胺的碰撞能为23V。
本发明第三方面涉及一种试剂盒,包含,
Ⅰ.盐酸水溶液;
II.氢氧化钠固体和/或氢氧化钠水溶液;和
III.乙醚和/或叔丁基甲醚。
在一个实施方案中,所述试剂盒还包含醋酸和甲醇;优选地,所述醋酸的纯度为99%-100%(W/W),更优选为99.5%(W/W)。
在一个实施方案中,所述试剂盒还包含液相色谱柱;优选地,所述液相色谱柱为菲罗门Agilent ZORBAX SB-C3液相色谱柱。
在一个实施方案中,所述试剂盒还包含0.1%(V/V)乙酸水溶液。
在一个实施方案中,所述盐酸水溶液的浓度为1-10%(W/W),优选为5%(W/W)。
本发明第四方面涉及本发明第三方面任一项所述试剂盒在提取和/或检测样品中的芳香胺中的用途;所述样品为烟草和/或烟草制品。
本发明第四方面的一个实施方式中,所述芳香胺选自邻氨基偶氮甲苯、4-氨基联苯、2-氨基-4-硝基甲苯、联苯胺、对氯苯胺、4-氯-邻甲基苯胺、2-甲氧基-5-甲基苯胺、2-奈胺、4,4'-二氨基二苯基甲烷、2,4-二氨基甲苯、3,3'-二氯联苯胺、3,3'-二甲氧基联苯胺、3,3'-甲基-4,4'-二氨基二苯甲烷、4,4'-亚甲基-二-(2-氯苯胺)、4,4'-二氨基二苯醚、邻甲苯胺、4-氨基偶氮苯、2-氨基苯甲醚、2,4-二甲基苯胺和2,6-二甲基苯胺中的一种或多种。
本发明中,
如无特别说明,“强酸”是指在水溶液中几乎能全部电离的酸类,具有强的酸性反应,例如盐酸HCl、硫酸H2SO4、硝酸HNO3、高氯酸HCl4等。
如无特别说明,“浸提”是指利用适当的溶媒和方法,从原料中将可溶性有效成分浸出的过程。
如无特比说明,“芳香胺”是指氨分子中部分或全部氢原子被苯基或其它芳香基取代的衍生物。例如苯胺、二苯胺、三苯胺等。芳香胺为高沸点的液体或低熔点的固体,具有特殊的气味,毒性大,一般难溶于水。
如无特别说明,“脂肪醚”是指两个一价脂肪烃基与一个氧原子连接的化合物,通式是R-O-R′,例如二甲醚CH3-O-CH3、二乙醚C2H5OC2H5等。
如无特别说明,“氮气吹干”是指将氮气吹入加热样品的表面进行样品浓缩或干燥,具有省时、操作方便、容易控制等特点。
如无特别说明,“弱酸”是指在水溶液中只能小部分电离的酸类,具有弱的酸性反应。例如碳酸H2CO3、氢硫酸H2S、硼酸H3BO3等。
如无特别说明,“烟草”是指茄科,烟草属(Nicotiana L.),一年生或多年生草本,嗜好类作物。本属约有60多个种,大多是野生种。主要栽培种有红花烟草和黄花烟草。二者均原产南美洲,都是自然产生的双二倍体。红花烟草又称普通烟草,是世界性商品生产用烟种。又因调制方法及种源、地区、栽培措施不同而形成多种类型:烤烟、白肋烟、马里兰烟、雪茄烟、熏烟、香料烟、晒红烟、晒黄烟等。每一类型中又有很多栽培品种。黄花烟草以前苏联和印度栽培较多,中国新疆、甘肃等地有少量栽培。本属中的花烟草(N.alata Link and Otta)和粉蓝烟草(N.glauca Graham)栽种供观赏。
如无特别说明,“烟草制品”是指全部或部分由烟叶为原材料生产的、供抽吸、吸吮、咀嚼或鼻吸的制品。所述烟草制品包括但不限于卷烟和嚼烟。
如无特别说明,“内标法”是指仪器分析中绘制标准曲线的一种方法。其原理是:将一定量的内标物质加入不同浓度级次的标准溶液中,然后求出待测成分和内标物的信号强度之比,作为纵轴;再求出待测成分的量或待测成分与内标物量的比值,作为横轴,绘制关系曲线。一般情况下,所用内标物质的物理、化学性质与待测成分相似,且能与待测成分分离。内标法可消除测量仪器,测量条件的变化,进样量变化等的影响,从而提高了分析精密度,广泛用于色谱分析和光谱分析。
本发明取得的有益效果:
1、本发明的提取方法能更大程度地提取出烟草和/或烟草制品中的芳香胺化合物,提取率更高。
2、本发明的检测方法能更为快速、准确地检测出烟草和/或烟草制品中的芳香胺化合物含量。
3、本发明的检测方法检出限低、灵敏度更高、重现性好、操作更为简便。
4、本发明检测方法中,采用菲罗门Agilent ZORBAX SB-C3液相色谱柱,能将20种芳香胺很好地分离,几乎不存在峰形拖尾问题。
附图说明
为了使本发明的内容更容易被清楚的理解,下面根据本发明的具体实施例并结合附图,对本发明作进一步详细的说明,其中
图1为本发明提取方法的流程图。
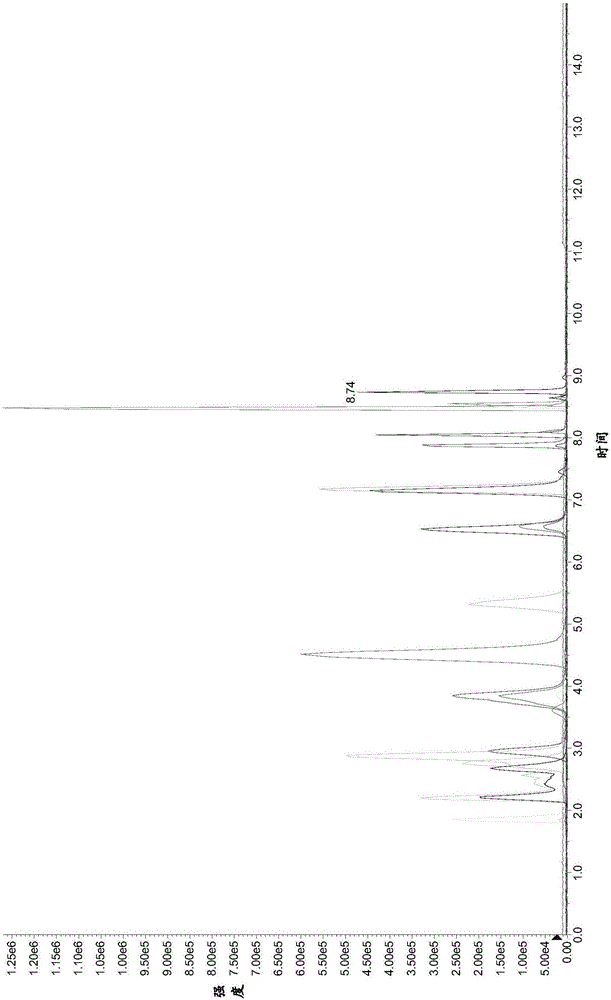
图2为本发明实施例1中S3级标准工作溶液的色谱图。
图3为对比例3中样品溶液的色谱图。
具体实施方式
实施例1
1、材料和试剂
(1)20种芳香胺的混合标准溶液(购买自百灵威科技有限公司):20种芳香胺分别为:邻氨基偶氮甲苯,4-氨基联苯,2-氨基-4-硝基甲苯,联苯胺,对氯苯胺,4-氯-邻甲基苯胺,2-甲氧基-5-甲基苯胺,2-奈胺,4,4'-二氨基二苯基甲烷,2,4-二氨基甲苯,3,3'-二氯联苯胺,3,3'-二甲氧基联苯胺,3,3'-甲基-4,4'-二氨基二苯甲烷,4,4'-亚甲基-二-(2-氯苯胺),4,4'-二氨基二苯醚,邻甲苯胺,4-氨基偶氮苯,2-氨基苯甲醚,2,4-二甲基苯胺,2,6-二甲基苯胺;浓度均为300μg/mL。
(2)内标物d7-1-氨基萘(购买自百灵威科技有限公司)。
(3)内标溶液:称取d7-1-氨基萘10mg,置于100mL容量瓶中,用甲醇稀释至刻度,配制成浓度为100μg/mL的一级内标储备溶液。取一级内标储备溶液1mL,置于100mL容量瓶中,用甲醇稀释至刻度,配制成浓度为1μg/mL的内标溶液。
(4)标准工作溶液:将20种芳香胺的混合标准溶液1mL,置于100mL棕色容量瓶中,以甲醇溶解和定容,得一级混合标准储备液(20种芳香胺的浓度均为3μg/mL)。取一级混合标准储备液5mL,置于100mL棕色容量瓶中,以甲醇溶解和定容,得二级混合标准储备液(20种芳香胺的浓度均为150ng/mL)。分别准确吸取二级混合标准储备液0.2mL、0.4mL、1mL、2mL、4mL和10mL于100mL棕色容量瓶中,以甲醇稀释至刻度,得到S1-S6级标准工作溶液,其中,20种芳香胺的浓度各自为0.3ng/mL、0.6ng/mL、1.5ng/mL、3ng/mL、6ng/mL和15ng/mL。
2、提取
按照图1的流程对烟草中的芳香胺进行提取。
准确称取0.5g烟末样品(粉碎后的云南烟叶),置于50mL磨口三角瓶中,准确加入50μL的1μg/mL的内标溶液和25mL5%(W/W)的盐酸水溶液,超声条件下浸提30分钟,得到初提液。之后用固体氢氧化钠将初提液的pH值调节至13,静置冷却5分钟后,加入10mL叔丁基甲醚振荡萃取10分钟,静置5分钟。向1mL上层液体中加入2至3滴醋酸并混合,氮气吹干后,向剩余物中加入1mL甲醇溶解,得到样品溶液。其中,醋酸的纯度为99.5%(W/W)。
3、检测
用高效液相色谱串联质谱仪(AB SCIEX公司的Qtrap 5500)分别对S1-S6级标准工作溶液和样品溶液进行检测分析。其中,S3级标准工作溶液的色谱图如图2所示。
色谱操作条件:色谱柱采用菲罗门Agilent ZORBAX SB-C3液相色谱柱,规格为150mm×2.1mm i.d.,5μm粒径;色谱柱温度为40℃;进样量为3μL;流动相:流动相A为甲醇,流动相B为0.1%(V/V)的乙酸水溶液;流动相的梯度洗脱程序见表1。
表1流动相的梯度洗脱程序
质谱操作条件:离子源:电喷雾电离源(ESI);扫描方式:正离子扫描;检测方式:多反应监测(MRM);电喷雾电压:5500V;离子源温度:650℃;辅助气Gas1压力:60psi;辅助气Gas2压力:50psi。
各芳香胺化合物的定量离子对、定性离子对及碰撞能量(CE)见表2。
表2质谱检测参数
4、计算
(1)根据上述3中S1-S6级标准工作溶液的检测结果,以芳香胺化合物与内标物的峰面积比对浓度比进行线性回归,得到20种芳香胺化合物的标准曲线方程,见表3。其中,横坐标表示芳香胺化合物的浓度与内标物浓度的比值,纵坐标表示芳香胺化合物的峰面积与内标物峰面积的比值。取最低浓度的标准工作溶液,进行10次平行测定,计算标准偏差,3倍标准偏差为检测限,10倍标准偏差为定量限,列于表3。
表3芳香胺化合物的标准曲线方程、检出限和定量限
由表3可知,本发明检测方法对20种芳香胺化合物的检出限低,灵敏度高。
(2)根据样品溶液的检测结果,结合表3中芳香胺化合物的标准曲线方程,计算出样品溶液中的芳香胺含量,进一步计算得到烟末样品中的芳香胺含量。在高、中、低三个水平上分别对20种芳香胺进行加标回收率测定,每个加标样品平行测定三次取平均值,并考察检测结果的重复性,结果如表4所示。
表4回收率和重复性
由表4可知,本发明检测方法对20种芳香胺的加标回收率都在90%以上,准确性高,而且检测结果的重复性良好。
对比例1
准确称取0.5g与实施例1同样的烟末样品,置于50mL磨口三角瓶中,准确加入50μL的1μg/mL的内标溶液和25mL5%(W/W)的盐酸水溶液,超声条件下浸提30分钟,得到初提液。将初提液经第一步SPE柱(Waters Oasis MCX)吸附,用1%HCL和甲醇洗脱除去中性和其它极性杂质,再用含5%NH4OH的甲醇洗脱。第二步SPE用非极性/疏水性反相柱(Waters Oasis HLB)吸附,洗脱液经浓缩后,进行LC-MS/MS分析,LC-MS/MS操作条件与实施例1相同。
结果显示:
1)对比例1测得的20种芳香胺化合物的色谱峰峰面积小于实施例1的测定结果,例如,对比例1测定的2-萘胺的峰面积为3746,而实施例1测定的2-萘胺的峰面积为6658,说明本发明提取方法的提取率更高,本发明检测方法的灵敏度更高。
2)对比例1中20种芳香胺的加标回收率均未超过90%,说明本发明检测方法相比于对比例1检测方法的准确性更高。
对比例2
用乙醚代替实施例1中的叔丁基甲醚,其余参照实施例1进行。
结果显示,与本发明叔丁基甲醚萃取得到的萃取液相比,使用乙醚萃取,得到的萃取液更混浊,杂质含量更多,污染性更大。
对比例3
用C18液相色谱柱代替实施例1的液相色谱柱,其余参照实施例1进行,得到样品溶液的色谱图如图3所示。
由图3可知,与本发明的液相色谱柱相比,使用C18液相色谱柱时,20种芳香胺不能很好地分离,并且峰形拖尾严重。
显然,上述实施例仅仅是为清楚地说明所作的举例,而并非对实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。而由此所引伸出的显而易见的变化或变动仍处于本发明创造的保护范围之中。
- 还没有人留言评论。精彩留言会获得点赞!