一种调味品中色素的检测方法与流程
本发明涉及一种色素的检测方法,尤其涉及一种调味品中色素的检测方法。
背景技术:
色素可以分为天然色素和合成色素。天然色素着色力和耐光性较差,因此合成色素更受人们的青睐。根据《食品添加剂使用标准(GB 2760-2014)》,我国仅允许使用柠檬黄、胭脂红等11种合成色素。然而合成色素种类繁多,一些不法商家为了增强着色效果,在调味品中添加我国明令禁止使用的色素,如苏丹红、罗丹明B、碱性橙等,这些违禁色素经人体代谢后可生成萘胺等致癌性物质,长期摄入会对人体造成损害。
违禁色素的检测方法主要包括高效液相色谱法和高效液相色谱串联质谱法。对于复杂基质样品,高效液相色谱法容易受到天然色素的干扰;而高效液相色谱串联质谱法特异性强,灵敏度高,受到的干扰较少。在高效液相色谱串联质谱法检测违禁色素方面,目前检测模式较为单一,通常只能采用正离子模式或者负离子模式进行检测,而且样品前处理过程复杂,需要耗费大量的时间。
技术实现要素:
基于此,本发明的目的在于克服上述现有技术的不足之处而提供一种前处理简单、能同时检测调味品中多达49种违禁色素、且检测准确度高的方法。
为实现上述目的,本发明采取的技术方案为:一种调味品中色素的检测方法,所述方法包括如下步骤:
(1)将样品进行前处理,得到上样液;
(2)采用UPLC-MS/MS方法对步骤(1)得到的上样液进行测定,并和标准溶液的测定结果进行比对分析;
其中,高效液相色谱条件为:
a)色谱柱:C18柱,100mm×3.0mm,粒径2.6μm;
b)流动相A为乙腈;流动相B为5~15mmol/L乙酸铵溶液;流速为0.3~0.7mL/min;洗脱方式:梯度洗脱;
质谱检测条件为:采用电喷雾离子源,正、负离子切换模式,电喷雾离子源温度为400~550℃;多反应监测模式;CAD碰撞气;气帘气压力为15~25psi;雾化气压力为50~60psi;辅助加热气压力为50~60psi。
优选地,所述高效液相色谱条件为:a)色谱柱:菲罗门KinetexC18柱,100mm×3.0mm,粒径2.6μm;b)流动相A为乙腈;流动相B为10mmol/L乙酸铵溶液;流速为0.5mL/min;洗脱方式:梯度洗脱;c)柱温:40℃;d)进样量:10μL;所述质谱检测条件为:采用电喷雾离子源,正、负离子切换模式,电喷雾离子源温度为500℃;多反应监测模式;气帘气压力:20psi;雾化气压力:55psi;辅助加热气压力:55psi;CAD碰撞气:高强度。
本发明所述UPLC-MS/MS方法高效液相色谱串联质谱法,具有高灵敏度和高选择性的优势,可以同时检测调味品中多种违禁色素。
优选地,所述前处理包括如下步骤:
(1)色素提取
按照每克调味品加入2.5mL提取剂的比例,将提取剂加至调味品中,混合均匀,离心,回收上清液;重复提取一次,合并提取液;
(2)QuEChERS方法净化提取液
向步骤(1)得到的提取液中加入C18吸附剂,再进行离心,取上清液用水稀释后过0.22μm有机滤膜,得到上样液。
更优选地,所述前处理包括如下步骤:
(1)色素提取
称取2g样品到50mL离心管中,加入5mL提取剂,漩涡混合均匀;将离心管以4500r/min的转速离心5min,回收上清液;重复提取一次,合并提取液;
(2)QuEChERS方法净化提取液
取1mL步骤(1)得到的提取液于2mL离心管中,加入吸附剂,涡旋1min,以8000r/min的转速离心2min,取0.2~0.4mL上清液,用水稀释至1mL,过0.22μm有机滤膜后,得到上样液。
本发明所述QuEChERS(Quick,Easy,Cheap,Effective,Rugged and Safe)方法快速、简便、成本低廉、易于操作。本发明方法采用改进的QuEChERS方法处理样品,使调味品中的49种违禁色素同时提取、净化,同时简化了样品处理步骤,具有高效、快速、成本低、操作简便等优势,净化效果好,灵敏度高,准确度和精密度均符合多残留分析方法的要求。
优选地,所述步骤(1)中的提取剂为乙腈和甲醇的混合物,所述提取剂中乙腈的体积百分比为20%~80%。
更优选地,所述提取剂中乙腈的体积百分比为50%。本发明所述提取剂,能同时兼顾极性和非极性色素,很好的对色素进行分离提取,提取率高,有利于提高检测的准确性。
优选地,所述步骤(2)中提取液用水稀释时,提取液和水的体积比为1:2.5~1:5。本发明按照所述比例对样品提取液进行稀释,可有效改善目标色素峰型,减少溶剂效应,有利于提高检测的准确性。
优选地,所述步骤(2)中的有机滤膜为聚四氟乙烯滤膜。聚四氟乙烯滤膜具有极强的化学兼容性,几乎能胜任所有的有机溶剂和强腐蚀化学品的过滤,并且不会对色素产生吸附。在本发明中,将提取液通过聚四氟乙烯滤膜进行过滤,能对提取液起到很好的净化效果。
优选地,所述调味品中的色素包括酸性蓝1、酸性橙II、碱性嫩黄O、碱性绿1、罗丹明B、结晶紫、苏丹橙G、苏丹黄、苏丹红7B、对位红、溶剂黄13、苏丹红I、苏丹红II、苏丹红III、苏丹红IV、二乙基黄、酸性红1、酸性红73、专利蓝V、橙黄IV、碱性橙21、硫黄素T、苏丹红G、分散橙37、分散橙1、分散红1、分散橙3、分散橙11、碱性红14、碱性蓝1、碱性紫2、碱性紫7、碱性橙22、酸性红52、酸性橙20、藏花橙G、酸性蓝3、酸性绿16、酸性绿A、溶剂黄1、溶剂黄3、溶剂橙2、甲基橙、萘基红、4-羟基偶氮苯、4-羟基-4-二甲氨基偶氮苯、罗丹明6G、分散黄3、柑橘红2。
优选地,以体积百分比计,所述梯度洗脱条件为:0~2.5min,10%~40%A;2.5~4.7min,40.0%A;4.7~7.0min,40%~90%A;7.0~12.0min,90%A;12.0~12.1min,10%A;12.1~17.0min,10%A;各洗脱时间段中均以流动相B补足余下的体积百分比。
本发明技术方案中流动相A为乙腈,流动相B为10mmol/L乙酸铵溶液,洗脱方式为梯度洗脱,流速为0.5mL/min;初始流动相比例为10:90(A:B体积比),到2.5min时流动相比例变为40:60,到7min时流动相比例变为90:10,12.1min时流动相比例又变为10:90,这个比例一直维持到17min,因此,本发明色谱柱和流动相的选择对调味品中违禁色素达到了优异的分离效果,进一步保证了本发明检测方法的准确性。
优选地,所述电喷雾离子源在0~4.1min为负离子模式,喷雾电压为-4500V;在4.1~16.0min为正离子模式,喷雾电压为5500V。
本发明采用正负离子两种不同的模式进行切换检测方法,同时对色素的阴、阳离子进行检测,解决了现在违禁色素检测模式单一、检测种类有限的技术难题,通用性强、检测对象覆盖范围宽,降低了漏检风险,缩短了检测周期。
相对于现有技术,本发明的有益效果为:
本发明采用液相色谱结合质谱法对调味品中多达49种违禁色素进行检测,采用正负离子切换模式,同时对色素的阴、阳离子进行检测,解决了现在违禁色素检测模式单一、检测种类有限的技术难题,通用性强、检测对象覆盖范围宽,具有高速、高效、高分辨、微量检测及分析自动化的性能和技术优势,降低了漏检风险;加上QuEChERS净化方法去除样品中的油脂,有效地改善了色素检测中复杂的前处理过程,简便快速,与以往多个方法的检测方法相比,分析时间短,灵敏度高,检测效率得到了根本性的提升,大大缩短了检测周期。
附图说明
图1为49种违禁色素标准品的总离子流图,其中0~4.1min为13种阴离子色素,4.1~17min为36种阳离子色素;
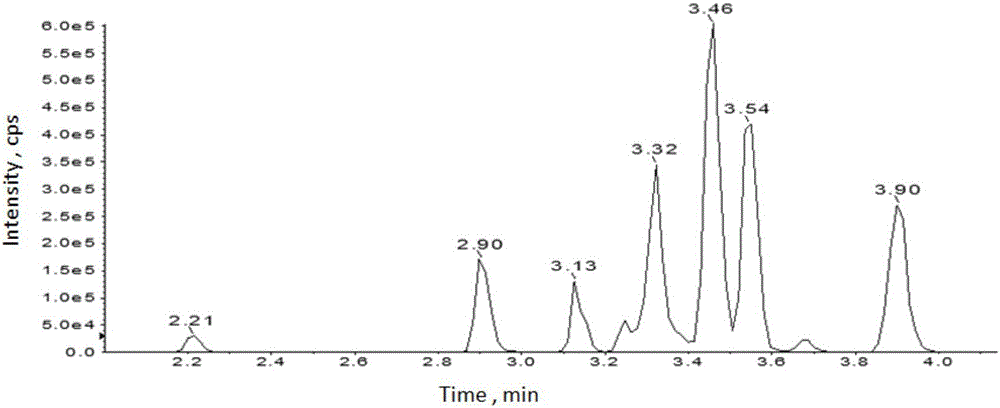
图2为13种阴离子色素负离子模式的提取离子流图,其中,各色素保留时间分别为:酸性红1:2.21min;酸性橙20:2.90min;酸性红73:3.13min;酸性绿16:3.25min;酸性蓝1:3.30min;甲基橙:3.32min;酸性绿A:3.37min;酸性红52:3.45min;藏花橙G:3.45min;酸性橙II:3.46min;专利蓝V:3.55min;酸性蓝3:3.55min;橙黄IV:3.90min;
图3为36种阳离子色素正离子模式的提取离子流图,其中,各色素保留时间分别为:碱性嫩黄O:4.88min;硫黄素T:4.89min;碱性紫2:5.00min;碱性红14:5.66min;碱性橙21:6.29min;罗丹明B:6.49min;苏丹橙G:6.51min;对羟基偶氮苯:6.73min;溶剂黄1:6.75min;溶剂黄13:6.82min;4-羟基-4-二甲氨基偶氮苯:6.82min;分散橙3:6.93min;分散黄3:6.97min;分散橙11:7.03min;碱性蓝1:7.07min;分散红1:7.22min;溶剂黄3:7.48min碱性紫7:7.52min;萘基红:7.53min;碱性橙22:7.56min;罗丹明6G:7.75min;对位红:7.84min;分散橙37:7.84min;苏丹黄:7.87min;柑橘红2:7.97min;分散橙1:7.99min;苏丹红G:8.05min;结晶紫:8.09min;苏丹红I:8.10min;二乙基黄:8.23min;溶剂橙2:8.38min;苏丹红II:8.77min;碱性绿1:9.06min;苏丹红III:9.31min;苏丹红7B:10.39min;苏丹红IV:10.64min;
图4为辣椒粉样品检出罗丹明B的总离子流图;
图5为辣椒粉样品检出罗丹明B的m/z的值为399.0的子离子提取流图;
图6为辣椒粉样品检出罗丹明B的m/z的值为355.0的子离子提取流图。
具体实施方式
为更好的说明本发明的目的、技术方案和优点,下面将结合附图和具体实施例对本发明作进一步说明。
实施例1
本发明所述调味品中色素的检测方法的一种实施例,本实施例调味品为一种调味酱,本实施例所述方法包括如下步骤:
(1)色素提取
称取2g样品到50mL离心管中,加入5mL提取剂,漩涡混合均匀;提取剂由乙腈和甲醇以50:50的体积比混合;将离心管以4500r/min的转速离心5min,回收上清液;重复提取一次,合并提取液。
(2)QuEChERS方法净化提取液
取1mL提取液于2mL离心管中,加入25mg C18吸附剂,涡旋1min,以8000r/min的转速离心2min。取0.3mL上清液,用水稀释至1mL,过0.22μm聚四氟乙烯滤膜后,待测定。
(3)UPLC-MS/MS分析
色谱柱:菲罗门Kinetex-C18(100mm×3.0mm,2.6μm);柱温:40℃;进样量:10μL;流动相A为乙腈;流动相B为10mmol/L乙酸铵溶液;流速为0.5mL/min;以体积百分比计,所述梯度洗脱条件为:0~2.5min,10%~40%A;2.5~4.7min,40.0%A;4.7~7.0min,40%~90%A;7.0~12.0min,90%A;12.0~12.1min,10%A;12.1~17.0min,10%A;各洗脱时间段中均以流动相B补足余下的体积百分比;
质谱条件:电喷雾离子源,0~4.1min为负离子模式(ESI-),喷雾电压-4500V;4.1~16.0min为正离子模式(ESI+),喷雾电压5500V;多反应监测模式(MRM);气帘气压力:20psi;雾化气(GS1)压力:55psi;辅助加热气(GS2)压力:55psi;CAD碰撞气:高强度;电喷雾离子源温度为500℃,具体见违禁色素的质谱参数表1所示。
表1 色素质谱检测参数
*定量离子对
按照上述条件,对49种违禁色素标准品和调味酱提取液进行UPLC-MS/MS分析和比对,对调味酱提取液中49种违禁色素进行定性定量分析。
(4)线性方程、相关系数、检出限及定量限
在UPLC-MS/MS分析时,按照常规方法绘制49种违禁色素的标准曲线,根据其标准曲线可以确定49种违禁色素线性方程、相关系数、检出限和定量限,其分析结果列于表2。
表2 49种违禁色素的线性方程、相关系数、检出限和定量限
(5)回收率及精密度
在调味品空白样品(调味酱)中,分别进行3组不同浓度水平的加标回收试验,每个水平作6次平行试验。其回收率及相对标准偏差见表3:
表3 49种违禁色素的回收率及相对标准偏差
由表3可知,各目标物回收率为70.98%~109.72%,相对标准偏差为0%~9.14%,所述方法显示了较好的回收率和精密度。
实施例2
本发明所述调味品中色素的检测方法的一种实施例,本实施例调味品为一种调味酱,本实施例所述方法包括如下步骤:
(1)色素提取
称取2g样品到50mL离心管中,加入5mL提取剂,漩涡混合均匀;提取剂由乙腈和甲醇以50:50的体积比混合;将离心管以4500r/min的转速离心5min,回收上清液;重复提取一次,合并提取液。
(2)QuEChERS方法净化提取液
取1mL提取液于2mL离心管中,加入25mg C18吸附剂,涡旋1min,以8000r/min的转速离心2min。取0.3mL上清液,用水稀释至1mL,过0.22μm聚四氟乙烯滤膜后,待测定。
(3)UPLC-MS/MS分析
色谱柱:菲罗门Kinetex-C18(100mm×3.0mm,2.6μm);柱温:40℃;进样量:10μL;流动相A为乙腈;流动相B为10mmol/L乙酸铵溶液;流速为0.5mL/min;以体积百分比计,所述梯度洗脱条件为:0~2.5min,10%~40%A;2.5~4.7min,40.0%A;4.7~7.0min,40%~90%A;7.0~12.0min,90%A;12.0~12.1min,10%A;12.1~17.0min,10%A;各洗脱时间段中均以流动相B补足余下的体积百分比;
质谱条件:电喷雾离子源,0~4.1min为负离子模式(ESI-),喷雾电压-4500V;4.1~16.0min为正离子模式(ESI+),喷雾电压5500V;多反应监测模式(MRM);气帘气压力:20psi;雾化气(GS1)压力:55psi;辅助加热气(GS2)压力:55psi;CAD碰撞气:高强度;电喷雾离子源温度为500℃,具体见违禁色素的质谱参数表1所示。
按照上述条件,对49种违禁色素标准品和调味酱提取液进行UPLC-MS/MS分析和比对,对调味酱提取液中49种违禁色素进行定性定量分析。
(4)线性方程、相关系数、检出限及定量限
在UPLC-MS/MS分析时,按照常规方法绘制49种违禁色素的标准曲线,根据其标准曲线可以确定49种违禁色素线性方程、相关系数、检出限和定量限。其分析结果列于表4。
表4 49种违禁色素的线性方程、相关系数、检出限和定量限
(5)回收率及精密度
在调味品空白样品(辣椒粉)中,分别进行3组不同浓度水平的加标回收试验,每个水平作6次平行试验,其回收率及相对标准偏差见表5。
表5 49种违禁色素的回收率及相对标准偏差
由表5可知,各目标物回收率为70.75%~114.68%,相对标准偏差为0.06%~9.38%,所述方法显示了较好的回收率和精密度。
实施例3
本发明所述调味品中色素的检测方法的一种实施例,本实施例调味品为一种调味酱,本实施例所述方法包括如下步骤:
(1)色素提取
称取2g样品到50mL离心管中,加入5mL提取剂,漩涡混合均匀;提取剂为乙腈和甲醇的混合物,其中乙腈的体积百分含量为20%;将离心管以4500r/min的转速离心5min,回收上清液;重复提取一次,合并提取液。
(2)QuEChERS方法净化提取液
取1mL提取液于2mL离心管中,加入25mg C18吸附剂,涡旋1min,以8000r/min的转速离心2min。取0.2mL上清液,用水稀释至0.7mL,过0.22μm聚四氟乙烯滤膜后,待测定。
(3)UPLC-MS/MS分析
色谱柱:菲罗门Kinetex-C18(100mm×3.0mm,2.6μm);柱温:40℃;进样量:10μL;流动相A为乙腈;流动相B为5mmol/L乙酸铵溶液;流速为0.3mL/min;以体积百分比计,所述梯度洗脱条件为:0~2.5min,10%~40%A;2.5~4.7min,40.0%A;4.7~7.0min,40%~90%A;7.0~12.0min,90%A;12.0~12.1min,10%A;12.1~17.0min,10%A;各洗脱时间段中均以流动相B补足余下的体积百分比;
质谱条件:电喷雾离子源,0~4.1min为负离子模式(ESI-),喷雾电压-4500V;4.1~16.0min为正离子模式(ESI+),喷雾电压5500V;多反应监测模式(MRM);气帘气压力:15psi;雾化气(GS1)压力:50psi;辅助加热气(GS2)压力:50psi;CAD碰撞气:高强度;电喷雾离子源温度为400℃,具体见违禁色素的质谱参数表1所示。
按照上述条件,对49种违禁色素标准品和调味酱提取液进行UPLC-MS/MS分析和比对,对调味酱提取液中49种违禁色素进行定性定量分析。
(4)线性方程、相关系数、检出限及定量限
根据实施例1和实施例2中同样的方法绘制49种违禁色素的标准曲线,根据其标准曲线可以确定49种违禁色素线性方程、相关系数、检出限和定量限。分析发现多数目标物的回收率在71.99%~109.22%范围内,相对标准偏差为0.07%~9.2%,所述方法显示了较好的回收率和精密度。
实施例4
本发明所述调味品中色素的检测方法的一种实施例,本实施例调味品为一种调味酱,本实施例所述方法包括如下步骤:
(1)色素提取
称取2g样品到50mL离心管中,加入5mL提取剂,漩涡混合均匀;提取剂为乙腈和甲醇的混合物,其中乙腈的体积百分含量为80%;将离心管以4500r/min的转速离心5min,回收上清液;重复提取一次,合并提取液。
(2)QuEChERS方法净化提取液
取1mL提取液于2mL离心管中,加入25mg C18吸附剂,涡旋1min,以8000r/min的转速离心2min。取0.4mL上清液,用水稀释至1mL,过0.22μm聚四氟乙烯滤膜后,待测定。
(3)UPLC-MS/MS分析
色谱柱:菲罗门Kinetex-C18(100mm×3.0mm,2.6μm);柱温:40℃;进样量:10μL;流动相A为乙腈;流动相B为15mmol/L乙酸铵溶液;流速为0.7mL/min;以体积百分比计,所述梯度洗脱条件为:0~2.5min,10%~40%A;2.5~4.7min,40.0%A;4.7~7.0min,40%~90%A;7.0~12.0min,90%A;12.0~12.1min,10%A;12.1~17.0min,10%A;各洗脱时间段中均以流动相B补足余下的体积百分比;
质谱条件:电喷雾离子源,0~4.1min为负离子模式(ESI-),喷雾电压-4500V;4.1~16.0min为正离子模式(ESI+),喷雾电压5500V;多反应监测模式(MRM);气帘气压力:25psi;雾化气(GS1)压力:60psi;辅助加热气(GS2)压力:60psi;CAD碰撞气:高强度;电喷雾离子源温度为550℃,具体见违禁色素的质谱参数表1所示。
按照上述条件,对49种违禁色素标准品和调味酱提取液进行UPLC-MS/MS分析和比对,对调味酱提取液中49种违禁色素进行定性定量分析。
(4)线性方程、相关系数、检出限及定量限
根据实施例1和实施例2中同样的方法绘制49种违禁色素的标准曲线,根据其标准曲线可以确定49种违禁色素线性方程、相关系数、检出限和定量限。分析发现多数目标物的回收率在68.33%~114.22%范围内,相对标准偏差为0.07%~9.4%,所述方法显示了较好的回收率和精密度。
实施例5
本实施例针对本发明提取方法中的提取剂的回收率进行了相关研究,采用对中间加标量进行加标回收试验,对3种不同比例的提取溶剂进行加标回收试验,得到3组不同的回收率并进行比较,加标浓度为表5中每种色素加标浓度的中间浓度,具体见表5中间的加标浓度点,得到回收率,研究结果如表6所示:
表6为不同体积比的乙腈与甲醇混合提取剂的加标回收率比较
从表6可以看出,本发明所述体积比的乙腈与甲醇混合提取剂,对样品中色素的提取效果较好,尤其是当提取剂中乙腈与甲醇的体积比为1:1时,对所有待测样品中色素的加标回收率相对较高(70.58%-109.16%)。
最后所应当说明的是,以上实施例仅用以说明本发明的技术方案而非对本发明保护范围的限制,尽管参照较佳实施例对本发明作了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的实质和范围。
- 还没有人留言评论。精彩留言会获得点赞!