病毒活性快速检测方法与流程
本发明涉及生物技术领域,尤其涉及一种病毒活性快速检测方法。
背景技术:
人类的很多疾病由病毒引起,目前有效的抗病毒性药物较少,病毒性疾病的防治主要需要依靠疫苗等进行预防。因此要控制病毒的传播、最小化病毒所致死亡率,对病毒活性进行快速、准确的检测至关重要。
病毒需要在宿主细胞内才能增殖,活病毒首先通过随机碰撞、静电引力,与宿主细胞形成非特异性吸附;然后病毒通过自身的粘附素与宿主细胞表面的特异性黏附受体结合,导致细胞膜发生改变,病毒核酸或病毒进入靶细胞内,进行生物合成、装配和释放。传统病毒检测方法是通过测定活病毒感染并杀死宿主细胞后形成的空斑数量(空斑形成单位,plaqueformingunit,pfu),测定是否存在活病毒,计算空斑形成单位(pfu)确定病毒滴度,并进一步对病毒进行分型。空斑形成实验是病毒活性的金标准,但由于通过观察病毒裂解细胞后形成的空斑检测病毒活性和病毒滴度,培养时间长,通常需要2-4天的时间,且干扰因素多,操作步骤繁多,需要一定熟练程度的人来操作,无法满足实际需要,更重要的是有些情况下病毒不能被培养出来。pcr和免疫荧光等方法可灵敏快速地测定靶病毒滴度和分型,但这些方法均无法测定病毒活性。因此,开发无需培养的病毒活性快速检测技术,在病毒性疾病的预防和控制中十分重要。
为了开发无需培养的病毒活性快速检测技术,本发明利用分子印迹聚合物(molecularlyimprintedpolymers,mips),目前,细菌的mips制备已有少量研究,对哺乳动物细胞的mips则极少有报道。在细菌mip中,部分研究者以细菌表面蛋白为模板制备mips,例如khan等人在单壁碳纳米管上,以金黄色葡萄球菌外层有代表性的蛋白a为模板,通过循环伏安法使3-氨基酚发生电聚合制备mips,在有牛血清白蛋白干扰的情况下,mips对模板仍有良好的选择性(khanmsr,moreiraftc,riujetal.plasticantibodyfortheelectrochemicaldetectionofbacterialsurfaceproteins.sensoractuatb-chem,2016,233:697-704.)。jiang等人在磁性纳米颗粒表面,以革兰阴性菌的信号分子ahls为模板,通过表面印迹制备了mips,可对细菌进行间接检测(jiangh,jiangd,shaojetal.magneticmolecularlyimprintedpolymernanoparticlesbasedelectrochemicalsensorforthemeasurementofgram-negativebacterialquorumsignalingmolecules(n-acyl-homoserine-lactones).biosensbioelectron,2016,75:411-419.)。idil等人以整个大肠杆菌为模板,以n-甲基丙烯酰-l-组氨酸、2-甲基丙烯酸羟乙酯为功能单体,以乙二醇二甲基丙烯酸为交联剂,用紫外光进行引发,制备大肠杆菌mips,可特异性吸附模板大肠杆菌。对大肠杆菌的检出限为70cfu/ml,检测范围为1.0×102-1.0×107cfu/ml。该方法用于果汁和河水中加标大肠杆菌的检测,回收率为81-97%(idiln,hedstromm,denizliaetal.wholecellbasedmicrocontactimprintedcapacitivebiosensorforthedetectionofescherichiacoli.biosensbioelectron,2017,87:807-815.)。roy等人以整个大肠杆菌为模板,以n,n’-亚甲基双丙烯酰胺为功能单体制备大肠杆菌mips。与亚铁/铁氰化物氧化还原探针连用,可实现对大肠杆菌的检测,且与大肠杆菌在10-109cfu/ml范围线性相关,检出限为10cfu/ml。除了能检测外,该材料还可从水样中捕获超过98%的大肠杆菌。最后该材料还可以通过光热在5分钟内杀死105cfu/ml大肠杆菌(roye,patras,tiwariaetal.singlecellimprintingonthesurfaceofag-znobimetallicnanoparticlemodifiedgrapheneoxidesheetsfortargeteddetection,removalandphotothermalkillingofe.coli.biosensbioelectron,2017,89(pt1):620-626.)。以上研究均表明细菌mips可特异性结合并检测靶细菌,但是,如何利用mips测定病毒活性至今仍是需亟待解决的问题。
技术实现要素:
为解决以上问题,本发明的目的是提供一种病毒活性快速检测方法,该检测方法具有快速、安全、准确的特点。
为实现上述目的,本发明所采用的技术方案是:
病毒活性快速检测方法,包括以下步骤:
1)制备宿主靶细胞的荧光分子印迹聚合物:
首先称取多巴胺和丹磺酰多巴胺,配制多巴胺/丹磺酰多巴胺混合溶液;然后将多巴胺/丹磺酰多巴胺混合溶液、宿主靶细胞与载体混合均匀,以宿主靶细胞为模板、丹磺酰多巴胺为荧光功能单体、多巴胺为功能单体,在载体表面进行聚合反应得到宿主靶细胞-丹磺酰多巴胺/多巴胺-载体复合体;最后利用洗脱剂将宿主靶细胞从宿主靶细胞-丹磺酰多巴胺/多巴胺-载体复合体上洗脱下来,得到宿主靶细胞的荧光分子印迹聚合物;
2)检测病毒活性:
向荧光分子印迹聚合物中加入宿主靶细胞和病毒样品,即刻测定荧光值为i0,待病毒样品中的病毒完全侵染宿主靶细胞后,测定荧光值为i,i/i0-1表示荧光变化水平,通过荧光变化水平确定病毒样品中是否含有靶病毒以及该靶病毒的含量。
作为优选方案,所述步骤1)中载体为经聚多巴胺修饰后的多巴胺-载体复合体,所述多巴胺-载体复合体是通过将多巴胺和载体材料混合,并在过硫酸铵催化作用下,多巴胺在载体材料表面发生自聚合反应而制得。载体材料经多巴胺修饰后得到的多巴胺-载体复合体能提供大量的氨基和儿茶酚基,可与细菌壁外层的功能基团发生作用,增加了与细菌的结合能力,从而使荧光变化更明显,使测定的结果更加准确。
作为优选方案,所述载体材料为纳米微球、微米微球或多孔板。
作为优选方案,所述多巴胺/丹磺酰多巴胺混合溶液中还包括催化剂,所述催化剂为过硫酸铵。
作为优选方案,所述多巴胺/丹磺酰多巴胺混合溶液中多巴胺、丹磺酰多巴胺、过硫酸铵的摩尔比为4:1:1。
作为优选方案,所述宿主靶细胞为大肠杆菌或鸡胚细胞,所述大肠杆菌的浓度为104~106cfu/ml,所述鸡胚细胞的浓度为104~106个/ml;所述洗脱剂为去离子水。
本发明制得的荧光分子印迹聚合物,包括载体材料以及聚合在载体材料表面的丹磺酰多巴胺/多巴胺荧光层,所述丹磺酰多巴胺/多巴胺荧光层具有特异性结合宿主靶细胞的印迹孔穴。其中,所述载体材料为纳米微球、微米微球或多孔板,所述印迹孔穴的直径为0.8~1.7μm。
所述荧光分子印迹聚合物是通过荧光分子印迹聚合物的制备方法制备而得,所述荧光分子印迹聚合物的制备方法为:首先制备多巴胺-载体复合体:将多巴胺与载体材料混合,并在过硫酸铵催化作用下,多巴胺在载体材料表面发生自聚合反应制得多巴胺-载体复合体;然后制备宿主靶细胞-丹磺酰多巴胺/多巴胺-载体复合体:首先称取多巴胺、丹磺酰多巴胺和过硫酸铵,配制多巴胺/丹磺酰多巴胺混合溶液;然后将多巴胺/丹磺酰多巴胺混合溶液、宿主靶细胞和多巴胺-载体复合体混合均匀,以宿主靶细胞为模板、丹磺酰多巴胺为荧光功能单体、多巴胺为功能单体,在多巴胺-载体复合体表面进行聚合反应得到宿主靶细胞-丹磺酰多巴胺/多巴胺-载体复合体;最后洗脱宿主靶细胞:利用去离子水将宿主靶细胞从宿主靶细胞-丹磺酰多巴胺/多巴胺-载体复合体上洗脱下来,得到该宿主靶细胞的荧光分子印迹聚合物。
本发明制得的荧光分子印迹聚合物在测定水中病毒含量以及区分水中活病毒和死病毒的应用。由于荧光分子印迹聚合物耐受水中其他病毒、离子、有机物的干扰,采用荧光分子印迹聚合物能准确的区分水中活病毒和死病毒以及测定水中的病毒含量。
本发明的病毒活性快速检测方法的原理是:
分子印迹聚合物(molecularlyimprintedpolymers,mips)是化学合成的高分子聚合物,通过特定的空间结构和化学键(包括共价键、离子键、氢键等)特异性识别和结合靶分子,其吸附特异性与天然抗体类似,但耐受有机溶剂、离子、酸碱、高温和高压。其中含荧光的mips,又称荧光分子印迹聚合物(fluorescentmolecularlyimprintedpolymers,fmips),不仅可以特异性结合模板分子,同时模板分子的结合还可以导致荧光mip发生荧光信号的改变(如荧光波长改变,或者荧光强度改变),根据荧光信号的改变,即可测定与fmips特异性结合的模板分子的含量。
结合图1所示,本发明以宿主靶细胞为模板、丹磺酰多巴胺作为荧光功能单体,多巴胺作为功能单体,在载体表面制备宿主靶细胞-丹磺酰多巴胺/多巴胺-载体复合体,然后再从复合体上将宿主靶细胞洗脱,即得到可特异性吸附宿主靶细胞的荧光分子印迹聚合物(fmips)。
当活的宿主靶细胞与fmips结合时,可导致fmips的荧光减弱,加入带检测的病毒样品,如果样品中存在活病毒,则活病毒会侵染结合在fmips上的宿主靶细胞,造成细胞破裂,并从fmips上脱落,从而使fmips荧光增强。通过比较病毒样品加样前后的荧光改变,即可快速测定样品中是否存在活病毒,并初步确定活病毒的数量。
本发明中采用的丹磺酰多巴胺的制备方法参阅中国发明专利(授权公告号cn103992252b授权公告日2016.06.22)公开的一种多巴胺衍生物及分子印迹聚合物制备方法和应用。
本发明的优点在于:
1,本发明通过使用丹磺酰多巴胺和多巴胺在载体表面进行聚合反应制备得到宿主靶细胞的fmips,由于丹磺酰多巴胺和多巴胺在水相条件下即可进行聚合反应,在水相条件下保证了宿主细胞的结构稳定性,而且聚多巴胺可为细菌和细胞印迹提供丰富的羟基,提高了fmips的印迹效果。
2,宿主靶细胞的fmips对宿主靶细胞具有特异性识别能力。fmips集特异性吸附和荧光检测于一体,不仅可以特异性吸附宿主靶细胞,还可以根据荧光的改变,直接快速测定其结合的宿主靶细胞数量。
3,利用宿主靶细胞的fmips,可以特异性检测侵染宿主靶细胞的活病毒的浓度,还可区分活病毒和死病毒,而且还能检测细菌病毒和人类病毒等多种病毒,无需培养病毒,能快速检测病毒活性。
附图说明
图1为本发明病毒活性快速检测方法的流程图;
图2a为使用经聚多巴胺修饰的多巴胺-载体复合体和不使用聚多巴胺修饰的多巴胺-载体复合体,所制备的fmips吸附宿主靶细胞前后的荧光变化水平图;
图2b为多巴胺/丹磺酰多巴胺混合溶液的过硫酸铵、多巴胺和丹磺酰多巴胺配比优化图;
图2c为使用不同洗脱液所制备的fmips吸附宿主靶细胞后的荧光变化水平图;
图2d为使用不同的宿主靶细胞浓度制备的fmips吸附宿主靶细胞后的荧光变化水平图;
图3a为实施例1制得的e.coli-fmips-ⅰ的电镜扫描图;
图3b为对比例1制得的fnip的电镜扫描图;
图3c为fmips膜的厚度的测量;
图3d为聚多巴胺修饰多孔板得到的多巴胺-载体复合体上的单层多巴胺膜(pda)厚度的测量;
图3e为单层多巴胺膜(pda)、e.coli-fmips-ⅰ和fnip膜的热重曲线;
图4a为e.coli-fmips-ⅰ对e.coli285的吸附等温曲线图;
图4b为e.coli-fmips-ⅰ吸附动力学曲线;
图4c为e.coli-fmips-ⅰ吸附e.coli285时荧光随时间变化趋势线图;
图4d为e.coli-fmips-ⅰ对活菌和死菌的识别能力图;
图5a为e.coli-fmips-ⅰ检测f2噬菌体的荧光变化时间趋势图;
图5b为e.coli-fmips-ⅰ检测t4噬菌体的荧光变化时间趋势图;
图5c为e.coli-fmips-ⅰ检测m13噬菌体的荧光变化时间趋势图;
图6a为不同大肠杆菌fmips对相应噬菌体的荧光响应图(p<0.05);
图6b为e.coli285-fmip对不同噬菌体的荧光响应图(p<0.05)
图6c为e.colitg1-fmip对不同噬菌体的荧光响应图(p<0.05);
图6d为e.colibl21-fmip对不同噬菌体的荧光响应图(p<0.05);
图7a为fmip对不同种类活噬菌体及灭活噬菌体的荧光响应图(p<0.05);
图7b为不同感染复数引起的荧光响应图(p<0.05);
图8a为鸡胚细胞-fmip的吸附等温曲线;
图8b为鸡胚细胞的吸附动力学曲线;
图8c为鸡胚细胞-fmips对活细胞和死细胞的荧光响应图;
图8d为鸡胚细胞-fmips检测流感病毒的荧光响应图;
图中各部件标号如下:宿主靶细胞(大肠杆菌)1、多巴胺2、丹磺酰多巴胺3、96孔酶标板4、大肠杆菌-丹磺酰多巴胺/多巴胺-载体复合体5、大肠杆菌的荧光分子印迹聚合物6、加入大肠杆菌后的荧光分子印迹聚合物7、病毒8、加入病毒后的荧光分子印迹聚合物9;其中,荧光值的大小为6>9>7。
具体实施方式
为解决现有技术中通过测定活病毒感染并杀死宿主细胞后形成的空斑数量的方法,存在培养时间、长干扰因素多、操作步骤繁多的问题,本发明提供一种病毒活性快速检测方法,本方法从制备病毒靶细胞的荧光分子印迹聚合物为设计思路的出发,通过以宿主细胞为模板、丹磺酰多巴胺作为荧光功能单体,多巴胺作为功能单体,在载体表面发生聚合反应,制备宿主靶细胞-丹磺酰多巴胺/多巴胺-载体材料复合体,再从复合体上将靶细胞洗脱,得到可特异性吸附宿主靶细胞的荧光分子印迹聚合物(fmips)来达到上述设计目的。以下通过具体的实施例来对本发明的病毒活性快速检测方法进行详细地说明。
实施例1制备大肠杆菌的荧光分子印迹聚合物ⅰ
制备大肠杆菌的荧光分子印迹聚合物ⅰ的方法,包括步骤:
1)试剂的配制:
多巴胺溶液ⅰ:过硫酸铵与多巴胺以2:1的摩尔比溶于10mmol/ltris-hcl缓冲溶液(tris-hcl缓冲溶液ph=8);
多巴胺溶液ⅱ:多巴胺溶于10mmol/ltris-hcl缓冲溶液(tris-hcl缓冲溶液ph=8);
多巴胺/丹磺酰多巴胺混合溶液:多巴胺:丹磺酰多巴胺:过硫酸铵摩尔比=4:1:1的摩尔比溶于10mmol/ltris-hcl缓冲溶液(tris-hcl缓冲溶液ph=8);
载体材料:以96孔酶标板作为载体材料;
宿主靶细胞:104~106cfu/ml的大肠杆菌菌液,大肠杆菌采用e.coli285;
洗脱液:去离子水;
2)多巴胺-载体复合体的制备(聚多巴胺修饰载体材料):
向在96孔酶标板中的每孔加300μl多巴胺溶液ⅰ,置于37℃的空气中96孔酶标板与多巴胺发生自聚合反应,反应24h后将孔中多巴胺溶液倒去,得到多巴胺-载体复合体;
3)大肠杆菌-丹磺酰多巴胺/多巴胺-载体复合体的制备:
向多巴胺-载体复合体中加入多巴胺/丹磺酰多巴胺混合溶液和宿主靶细胞(大肠杆菌),置于摇床中(温度为37℃,转速为150rpm)缓慢振摇72h后倒去孔中的液体,即得大肠杆菌-丹磺酰多巴胺/多巴胺-载体复合体;
4)洗脱宿主靶细胞:
用去离子水反复洗涤大肠杆菌-丹磺酰多巴胺/多巴胺-载体复合体,将大肠杆菌-丹磺酰多巴胺/多巴胺-载体复合体中的大肠杆菌洗脱下来,每次洗涤30min,共洗涤6次,得到大肠杆菌的荧光分子印迹聚合物ⅰ,大肠杆菌的荧光分子印迹聚合物ⅰ以下均采用简称e.coli-fmips-ⅰ。
实施例2制备大肠杆菌的荧光分子印迹聚合物ⅱ
实施例2的制备方法与是实施例相比,区别在于:实施例2不进行实施例1中的步骤2),即实施例2不采用经聚多巴胺修饰的多巴胺-载体复合体作为载体,而是直接将多巴胺,丹磺酰多巴胺,大肠杆菌在载体材料上聚合。具体过程如下:
1)大肠杆菌-丹磺酰多巴胺/多巴胺-载体复合体的制备:向96孔酶标板中加入多巴胺/丹磺酰多巴胺混合溶液和宿主靶细胞(大肠杆菌),置于摇床中(温度为37℃,转速为150rpm)缓慢振摇72h后倒去孔中的液体,即得大肠杆菌-丹磺酰多巴胺/多巴胺-载体复合体;;
2)洗脱宿主靶细胞:用洗脱剂反复洗涤大肠杆菌-丹磺酰多巴胺/多巴胺-载体复合体,将大肠杆菌-丹磺酰多巴胺/多巴胺-载体复合体中的大肠杆菌洗脱下来,每次洗涤30min,共洗涤6次,得到大肠杆菌的荧光分子印迹聚合物ⅱ。
实施例3制备大肠杆菌的荧光分子印迹聚合物ⅲ
大肠杆菌的荧光分子印迹聚合物ⅲ的制备方法与实施例1相同,不同之处在于:本实施例将实施例1步骤2)中的多巴胺溶液ⅰ改为多巴胺溶液ⅱ,多巴胺溶液ⅰ与多巴胺溶液ⅱ的区别在于过硫酸铵的是否添加。本实施例制备的大肠杆菌的荧光分子印迹聚合物ⅲ的方法在此不再赘述。
实施例4制备鸡胚细胞的荧光分子印迹聚合物
鸡胚细胞的荧光分子印迹聚合物制备方法与实施例1相同,不同之处在于:本实施例将实施例1步骤3)中的宿主靶细胞改为鸡胚细胞(cek),本实施例制得的鸡胚细胞的荧光分子印迹聚合物以下均采用简称鸡胚细胞-fmips。
实施例5制备大肠杆菌的荧光分子印迹聚合物ⅳ
1)试剂的配制:
多巴胺溶液ⅰ:过硫酸铵与多巴胺以2:1的摩尔比溶于10mmol/ltris-hcl缓冲溶液(tris-hcl缓冲溶液ph=8);
多巴胺/丹磺酰多巴胺混合溶液:多巴胺:丹磺酰多巴胺:过硫酸铵摩尔比=4:1:1的摩尔比溶于10mmol/ltris-hcl缓冲溶液(tris-hcl缓冲溶液ph=8);
载体材料:以150mg/ml硅胶微球为载体材料;
宿主靶细胞:104~106cfu/ml的大肠杆菌菌液,大肠杆菌采用e.coli285;
洗脱液:去离子水
2)多巴胺-载体复合体的制备:
将150mg/ml硅胶微球与多巴胺溶液ⅰ在37℃搅拌混合10小时,硅胶微球与多巴胺发生自聚合反应,10小时后取出硅胶微球,得到多巴胺-载体复合体;
3)大肠杆菌-丹磺酰多巴胺/多巴胺-载体复合体的制备:
向多巴胺-载体复合体中加入多巴胺/丹磺酰多巴胺混合溶液和宿主靶细胞(大肠杆菌e.coli285),置于摇床中(温度为37℃,转速为150rpm)缓慢搅拌10小时后取出硅胶微球,即得大肠杆菌-丹磺酰多巴胺/多巴胺-载体复合体;
4)洗脱宿主靶细胞:
用洗脱剂反复洗涤大肠杆菌-丹磺酰多巴胺/多巴胺-载体复合体,将大肠杆菌-丹磺酰多巴胺/多巴胺-载体复合体中的大肠杆菌洗脱下来,每次洗涤30min,共洗涤6次,得到大肠杆菌的荧光分子印迹聚合物ⅳ。
实施例6制备大肠杆菌的荧光分子印迹聚合物ⅴ
大肠杆菌的荧光分子印迹聚合物ⅴ制备方法与实施例5相同,不同之处在于:本实施例将实施例5中的硅胶微球载体改为1mg/ml四氧化三铁纳米微球。
实施例7空白印迹聚合物(nip)的制备
分别制备实施例1~6的对比例,分别为对比例1、对比例2、对比例3、对比例4、对比例5和对比例6。对比例1、对比例2、对比例3、对比例4、对比例5和对比例6与对应的实施例的制备方法相同,区别在于:不添加宿主靶细胞。以对比例1为例,其他在此不在赘述。
对比例1的制备方法与实施例1的制备方法相同,区别在于:步骤3)中不添加宿主靶细胞(大肠杆菌)。对比例1~6制得的空白印迹聚合物以下以及说明说附图中均使用简称fnip。
实施例8反应参数的优化试验
向实施例1~6中制备的细菌或细胞的荧光分子印迹聚合物(fmips)以及各实施例对应的空白印迹聚合物(fnips)中,分别加入300μlpbs缓冲溶液,测其荧光值作为i0,倒掉pbs缓冲溶液后,加入模板菌液或细胞液300μl后测其荧光值,作为i,用i0/i-1表示荧光变化程度。每个浓度平行测定3次取平均值,结果分别见图2a、图2b、图2c、图2d。以下分别对图2a、图2b、图2c和图2d作详细的说明。
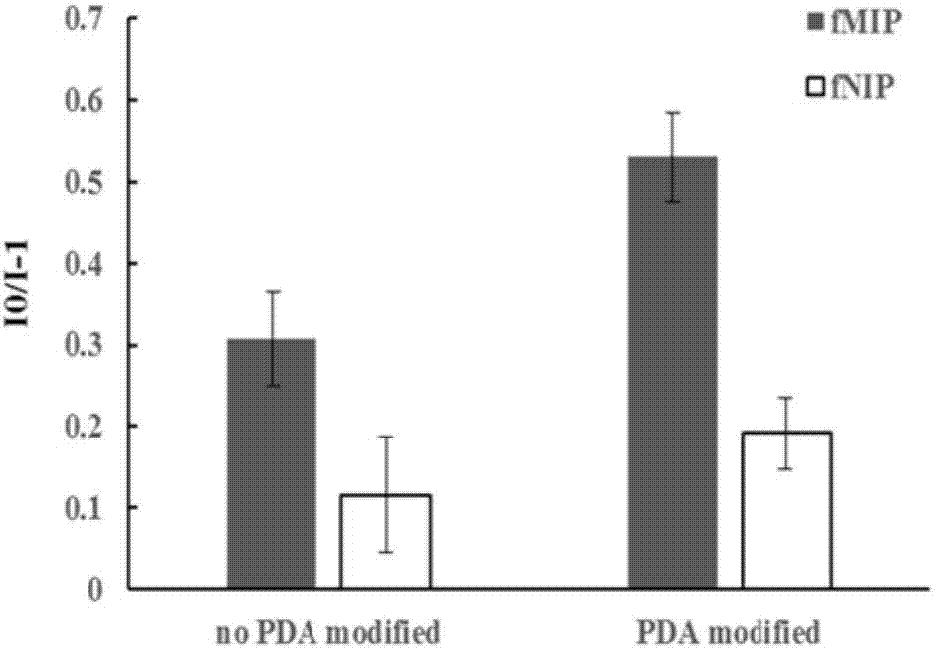
图2a为实施例1和实施例2的荧光变化水平对比图,即使用聚多巴胺修饰的载体材料和不使用聚多巴胺的载体材料所制备的fmips吸附宿主靶细胞前后的荧光变化水平图;实施例1中使用载体为聚多巴胺修饰的多巴胺-载体复合体,实施例2未对载体进行修饰;分别测定fmips吸附宿主靶细胞前后的荧光变化水平。由图2a可见,修饰在载体上的聚多巴胺膜(pda)的孔上再合成mips,荧光变化效果优于未修饰聚多巴胺膜的。其原理是:聚多巴胺膜能提供大量的氨基和儿茶酚基,可与细菌壁外层的功能基团发生作用,增加了与细菌的结合能力,从而使荧光变化明显。
图2b为多巴胺/丹磺酰多巴胺混合溶液的过硫酸铵、多巴胺和丹磺酰多巴胺配比优化图;通过测定过硫酸铵、多巴胺和丹磺酰多巴胺,在不同的摩尔比之下制备的fmip和fnip吸附靶细菌前后的荧光变化水平,选择最佳的配比。图2b可以看出,使用了过硫酸铵的mip荧光变化量提高了两倍以上,可知催化剂过硫酸铵的使用是成功制备抗病毒mip的前提。多巴胺:丹磺酰多巴胺与过硫酸铵的比例选为4:1:1时,fmip的荧光变化量最大,表明该条件下所制备的fmips吸附性能最佳。
图2c为使用不同洗脱液,所制备的fmips吸附宿主靶细胞后的荧光变化水平图;由图2c可以看出,以去离子水为洗脱液的效果明显优于3%乙酸与1mol/lnacl的混合液。其原理是在低渗环境中,细菌可因吸水过度而膨胀死亡。本实验中以去离子水为洗脱液,提供低渗环境,可使大肠杆菌发生死亡而洗脱下来。
图2d为使用不同的宿主靶细胞浓度时,所制备的fmips吸附宿主靶细胞后的荧光变化水平图;由图2d可以看出,当e.coli285菌悬液浓度为105cfu/ml时,荧光变化最为明显,在小于105cfu/ml时,随着模板滴度增加,能与模板结合的功能单体数量也增加,致使结合位点数量增加,荧光变化增加。
实施例9理化性质分析试验
图3a为实施例1制得的e.coli-fmips-ⅰ的电镜扫描图;
图3b为对比例1制得的fnip的电镜扫描图;
由图3a和图3b可看出,e.coli-fmips-ⅰ与fnip均为膜状物,图3a可见,大肠杆菌-丹磺酰多巴胺/多巴胺-载体复合体在洗脱完宿主靶细胞后存在形态、大小不一的孔穴,这些孔穴为大肠杆菌去除后留下的三维孔穴结构,形状有圆形、椭圆形,为大肠杆菌的横断面、不同侧面留下的印迹。孔穴直径为0.8~1.7μm,这与大肠杆菌的大小一致。图3b的fnips膜并没有看出明显的孔穴。
在以硅胶微球为载体材料,制备多巴胺-载体复合体,对多巴胺-载体复合体上的单层聚多巴胺膜(pda)、大肠杆菌285-fmips和fnips膜用元素分析仪对n、c、h元素含量进行分析,结果见表1。从表1可以看出,fmips、fnips膜中n、c元素含量相比单层pda膜明显增加,说明成功地在pda单层膜上制备了含更多n元素的丹磺酰多巴胺和多巴胺的fmips、fnips。
表1elementcompositionofpda,fmips,fnipsmembrane
图3c为fmips膜的厚度的测量;
图3d为修饰在载体上的单层多巴胺膜厚度的测量;
由图3c和图3d可以看出,fmips膜的厚度并不均一,范围从16.0~48.0μm,而且明显厚于单层多巴胺膜(约为2.8μm)。
图3e为单层多巴胺膜(pda)、e.coli-fmips-ⅰ和fnip膜的热重曲线;不同物质会随着温度不断升高而发生分解,导致重量下降,通过热重分析可判断形成的薄膜量。多巴胺在248℃开始分解,而交联后形成的聚多巴胺比较耐热,在400℃开始分解。从图3e中可以看出,从425℃开始三条线的重量有明显的下降,该温度正是聚多巴胺开始分解的温度,说明三种膜均有聚多巴胺。至700℃单层pda膜失重约0.4%,fmip膜约失重2%,fnip膜约失重1.3%,说明fmipf和nip膜明显厚于单层pda膜,成功在单层pda膜上制备了fmip。
实施例10e.coli-fmips-ⅰ对e.coli285的吸附特性
如图4a,e.coli-fmips-ⅰ对e.coli285的吸附等温曲线图;配制不同浓度的e.coli285菌悬液,分别加入到e.coli-fmips-ⅰ和对应的fnip中,吸附平衡后,用平板涂布法测定上清液中细菌的滴度。以吸附量adsorption(cfu/cm2)为纵坐标,菌液起始滴度c0(cfu/ml)为横坐标,绘制吸附等温线。从图4a可见当起始菌液滴度小于800cfu/ml时,吸附容量随着菌液的浓度增加而增加,大于800cfu/ml时,曲线趋于平缓,说明此时fmip上的识别位点均已与大肠杆菌模板结合,达到吸附饱和状态。
图4b为e.coli-fmips-ⅰ吸附动力学曲线;将100cfu/ml的e.coli285加入到fmip和fnip中,分别在0.5h、1h、2h、3h、6h、9h、12h时间点用平板涂布法测上清菌液滴度,同时测其荧光变化。以吸附量adsorption(cfu/cm2)为纵坐标,时间(h)为横坐标,绘制吸附动力学曲线;图4b显示,出在前1个小时,随时间增加,吸附量明显增加,吸附速率较快,吸附在1h后达到平衡。
图4c为e.coli-fmips-ⅰ吸附e.coli285时荧光随时间变化趋势线图;将100cfu/ml的e.coli285加入到fmip和fnip中,分别在0.5h、1h、2h、3h、6h、9h、12h时间点测定fmips和fnips的荧光;图4c显示,fmips和fnips的荧光,与吸附率对应,均在吸附1h后达到平衡。显示荧光变化量可以准确反映宿主细胞的吸附率。
图4d为e.coli-fmips-ⅰ对活菌和死菌的识别能力图;为评价e.coli-fmips-ⅰ在检测过程中是否能区分活菌与死菌,分别在fmips、fnips中加入e.coli285活菌、高压灭活菌、酒精灭活菌,待吸附平衡后,测定荧光变化i0/i-1。图4d显示fmips可以特异性识别活菌,对不同方法灭活的宿主细菌均可以有效识别。
实施例11e.coli-fmips-ⅰ检测细菌病毒的时间变化规律
图5a为e.coli-fmips-ⅰ检测f2噬菌体的荧光变化时间趋势图;
图5b为e.coli-fmips-ⅰ检测t4噬菌体的荧光变化时间趋势图;
图5c为e.coli-fmips-ⅰ检测m13噬菌体的荧光变化时间趋势图;
f2和t4噬菌体为烈性噬菌体,可使宿主菌发生裂解,图5a显示f2噬菌体在前0.5h能使荧光快速发生变化,在2h处达到平衡状态;图5b可以看出t4噬菌体在侵染宿主菌后的前0.5h使荧光变化最快,但在1h时达到平衡。其原理是与f2的裂解周期比t4长有关联。m13为温和噬菌体,可在不导致细菌死亡的情况下进行增殖,图5c显示m13噬菌体在刚侵染宿主菌后,fmip和fnip的荧光随时间没有很明显的变化,且fmip和fnip之间也没有明显差异。但在6~12小时范围内,mip荧光变化小幅度增加,其原理是吸附在fmips上的宿主菌开始逐渐死亡。以上结果说明本方法更适合用于可裂解宿主细胞的烈性病毒的快速检测。
实施例12利用不同大肠杆菌的fmip检测病毒活性
分别以e.coli285、e.colitg1、e.colibl21为宿主靶细胞,制备三种fmip,分别为285-fmip、tg1-fmip、bl21-fmip;分别加入f2、m13、t4的活噬菌体和灭活噬菌体去侵染相应的宿主菌。
图6a为不同大肠杆菌fmips对相应噬菌体的荧光响应(p<0.05);图6a可见,与活性噬菌体相比,灭活噬菌体没有引起fmips的明显响应。说明本方法对活性噬菌体和灭活噬菌体有很好的特异性,可用于病毒活性的检测。
图6b为e.coli285-fmip对不同噬菌体的荧光响应(p<0.05);在吸附了相应宿主菌的大肠杆菌285、tg1、bl21三种fmips中,每种fmip均加入f2、m13、t4三种噬菌体,分析该方法对不同大肠杆菌病毒的检测特异性。图6b显示,f2和t4噬菌体均可以侵染大肠杆菌285(f2噬菌体的宿主菌),引起285-fmip的荧光变化,即大肠杆菌285-fmip可以测定f2和t4噬菌体的活性。
图6c为e.colitg1-fmip对不同噬菌体的荧光响应(p<0.05);图6c显示f2和m13噬菌体可引起tg1-fmip荧光发生明显改变(tg1为m13的宿主菌),因m13为温和噬菌体,引起tg1-fmip荧光变化低于f2噬菌体,表明tg1-fmips可以同时测定f2和m13活性。
图6d为e.colibl21-fmip对不同噬菌体的荧光响应(p<0.05);图6d可以看出只有t4噬菌能引起bl21-fmip(t4噬菌体宿主菌)的荧光变化,f2、m13不能引起bl21-fmip荧光变化。可见,大肠杆菌fmip可用于对该菌有侵染性的噬菌体活性检测,若噬菌体不能侵染该菌则不会产生明显的荧光变化,证明此方法的特异性良好。
实施例13不同大肠杆菌的fmip病毒感染能力的测定
在吸附了相应宿主菌的285-fmip、tg1-fmip、bl21-fmip中,分别加入活的或灭活的f2、m13、t4活噬菌体;
图7a为fmip对不同种类活噬菌体及灭活噬菌体的荧光响应(p<0.05);图7a显示活性噬菌体可导致fmips荧光发生明显改变,而灭活噬菌体则不能引起fmip的明显响应。该方法对活性噬菌体和灭活噬菌体有很好的特异性,可用于病毒活性的检测。
图7b为不同感染复数引起的荧光响应(p<0.05),分析不同的感染复数(moi)对fmips荧光改变的影响,图7b可看出当moi≥1时,即可导致fmips的荧光发生明显的改变,差异经t检验,p<0.05,有统计学意义。说明1个噬菌体侵染1个大肠杆菌后,可在大肠杆菌细胞内繁殖引起大肠杆菌破裂死亡,使结合fmip的大肠杆菌数量减少,引起明显的荧光变化。这一数值明显低于病毒活性金标准:噬菌斑形成实验(moi=20),表明本方法检测病毒活性的灵敏度远高于噬菌斑形成实验法。
实施例14fmips检测人类病毒活性
为探讨本方法在检测人类病毒上的应用,首先利用优化的fmips制备条件,以鸡胚cek细胞为模板,制备了可特异性吸附鸡胚细胞-fmips。配制不同浓度的cek细胞悬液,分别加入到鸡胚细胞fmip和fnip中,吸附平衡后,用mtt法测定上清液中cek细胞的滴度,并绘制吸附等温线。从图8a可见当起始细胞滴度小于360个/ml时,吸附容量随着细胞液的浓度增加而增加,大于360个/ml时,曲线趋于平缓,说明此时鸡胚细胞-fmip上的识别位点均已与cek细胞板结合,达到吸附饱和状态。
将100个/ml的鸡胚cek细胞加入到鸡胚细胞-fmip和对应的fnip中,分别在0.5h、1h、2h、3h、6h、9h、12h时间点用mtt法测上清细胞滴度,绘制吸附动力学曲线。图8b显示,在1个小时内,随时间增加,吸附量明显增加,吸附速率较快,吸附在1h后达到平衡。
分别在fmips、fnips中加入活鸡胚cek细胞、高压灭活鸡胚cek细胞、酒精灭活鸡胚cek细胞,待吸附平衡后,测定荧光变化i0/i-1。图8c显示鸡胚细胞-fmips可以特异性识别活细胞,对不同方法灭活的宿主细胞均可以有效识别。
利用cek细胞制备的鸡胚细胞-fmips,检测了活的流感病毒和死的流感病毒,结果(图8d)显示鸡胚细胞-fmips可以特异性检测活流感病毒,对死病毒无明显的荧光改变。
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对本发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
- 还没有人留言评论。精彩留言会获得点赞!