白藜芦醇微胶囊的制备方法与流程
本发明属于食品工程技术领域,具体涉及一种以坛紫菜多糖复合卵清蛋白作为壁材制备白藜芦醇微胶囊的方法。
背景技术:
药物或生物活性物质在摄入机体后,只有尽可能让药物全部作用于病灶,才能最大程度发挥药物或者生物活性物质的效用,但是由于机体内部环境较为复杂多变,药物在达到目的部位之前可能会因为机体ph、温度、酶等因素发生降解或失活,因此对药物或者生物活性物质赋予一个合适的递送系统,让药物在到达需要作用的部位时仍然保持应有的活性和稳定,或者让它们在作用位点处拥有一定程度上的缓释性,对最大程度发挥药物或生物活性物质的效用具有十分重要的意义。
一直以来,微胶囊在递送药物或者活性物质方面被广泛研究和应用,微胶囊是一类尺寸从几微米到数百微米的微型容器,外壳材料的成分和芯材的选择很大程度上会影响微胶囊的性能,不同的外壳材料和芯材,它们的包封率、流动性、溶解性、渗透性、缓释性和其他性质都有所不同。高分子材料常常被选择用来作为微胶囊外壳的材料,其中多糖、蛋白质、固体脂和脂质体等天然高分子材料因为其毒性低、安全、包埋率高,且具有良好的生物相容性和生物降解性而被广泛研究。
白藜芦醇具有广泛的生物活性,如抗肿瘤、抗炎、抗氧化、抗病毒等,在治疗糖尿病、心血管疾病、神经衰退、心脏保护以及延长寿命方面有积极作用。虽然白藜芦醇在临床研究方面取得了可喜的成果,但是它在药物制剂的实际应用中仍然受到很大的限制,主要是因为白藜芦醇自身极差的水溶性和化学不稳定性,阻碍了其在人体内的吸收。另外,白藜芦醇摄入人体后会在胃肠道中经历了一个大的ⅱ相代谢过程,这与上述因素一起,导致了低效的全身给药,最终导致白藜芦醇在人体内的生物利用度降低,阻碍了其巨大的治疗潜力,因此就需要对其设计一个合理有效的药物递送系统。随着纳米颗粒在药物制剂领域的兴起,利用微胶囊技术包埋白藜芦醇的药物递送系统也越来越多地被人们探索。
技术实现要素:
本发明要解决的技术问题是提供一种制备白藜芦醇微胶囊的方法,采用本发明的方法不但能改善白藜芦醇的水溶性并增强其化学稳定性。
为了解决上述技术问题,本发明提供一种白藜芦醇微胶囊的制备方法,包括以下步骤:
1)、坛紫菜多糖分离纯化组分的制备:
用纤维素de-52离子交换层析柱和葡聚糖sephadexg-100凝胶层析柱对坛紫菜多糖进行分离和纯化,得到纯化坛紫菜多糖组分php2;
2)、白藜芦醇微胶囊的制备,包括以下步骤:
①、将卵清蛋白(ova)水溶液和php2水溶液分别置于(4±1)℃平衡(24±2)h;分别得平衡后ova溶液和平衡后php2溶液;
②、将白藜芦醇溶于乙醇,得白藜芦醇乙醇溶液;
③、将白藜芦醇乙醇溶液加入到平衡后ova溶液中超声(超声时间约为15~25min),然后旋转蒸发(用旋转蒸发仪)除去乙醇,再加入平衡后php2溶液,形成复合溶液;所述复合溶液中,白藜芦醇的浓度为(15±2)mg/l,ova和php2的质量之比为1:1,且ova和php2的浓度之和为(10±1)g/l;
将复合溶液加热至(60±10)℃磁力搅拌(搅拌时间约为25~35min),调节ph值3~4(优选ph为3.4)后震荡(震荡时间约为5~15min),置于(4±1)℃平衡(24±2)h;离心(10000rpm,20min),离心所得的沉淀进行洗涤和干燥(真空冷冻干燥36h),得白藜芦醇微胶囊(opr)。
作为本发明的白藜芦醇微胶囊的制备方法的改进,步骤1):
用于纤维素de-52离子交换层析柱的洗脱剂为0.5m的nacl溶液;
用于葡聚糖sephadex-100的洗脱液为去离子水。
作为本发明的白藜芦醇微胶囊的制备方法的进一步改进,步骤1)中:
依次以去离子水、0.1m、0.3m、0.5m的nacl溶液为洗脱剂对坛紫菜多糖(cphp)进行洗脱,控制流速1ml/min,
合并0.5mnacl溶液对应的洗脱液。
作为本发明的白藜芦醇微胶囊的制备方法的进一步改进,步骤2)的①中,卵清蛋白(ova)水溶液中卵清蛋白(ova)浓度为(1±0.1)g/100ml,php2水溶液中php2浓度为(1±0.1)g/100ml。
作为本发明的白藜芦醇微胶囊的制备方法的进一步改进,步骤2)的③中,
用hcl(1mhcl溶液)调节ph值3~4(优选ph为3.4)后,漩涡振荡器震荡(10±2)min;
用ph值3~4的hcl溶液对离心所得的沉淀进行洗涤。
本发明所得到的坛紫菜多糖组分php2,分子量为523kda,总糖含量66.89%,硫酸根含量8.33%,3,6-内醚半乳糖含量5.6%,蛋白质含量0.21%。单糖组成主要为半乳糖(99.03%),以及少量的甘露糖(0.26%)、鼠李糖(0.2%)、葡萄糖(0.1%)等。
本发明最终得到的微胶囊中,白藜芦醇的包埋率达95.4%,载药率达2.89%。
本发明坛紫菜多糖按文献(李银停.坛紫菜酶解多糖的分离纯化及生物活性研究[d].浙江工商大学,2019.)报道方法制备而得。
本发明具有如下技术优势:
(1)以坛紫菜多糖-卵清蛋白作为壁材制备白藜芦醇微胶囊,能够提高传统包埋方法的包埋率,白藜芦醇的包埋率可达95.4%。
(2)不仅能解决白藜芦醇的水溶性差的问题,而且能保护白藜芦醇,增强其的ph、紫外光分解和贮藏稳定性。
白藜芦醇微胶囊在中性或者弱酸性条件下以及14天避光贮藏条件下,粒径较小且均一,未出现续集沉淀等不溶现象,因此水溶性较佳;
经紫外光照射,微胶囊中的白藜芦醇相比较游离的白藜芦醇,能有效避免紫外光照射的分解,因此化学稳定性好。
(3)经包埋的白藜芦醇微胶囊,其对dpph自由基的清除率高于未包埋的白藜芦醇,且微胶囊具有显著的抗肿瘤活性,能有效抑制人宫颈癌细胞和人肝癌细胞的增殖。
白藜芦醇微胶囊可应用于药物制剂或者保健食品,有效提高白藜芦醇的生物利用率。
附图说明
下面结合附图对本发明的具体实施方式作进一步详细说明。
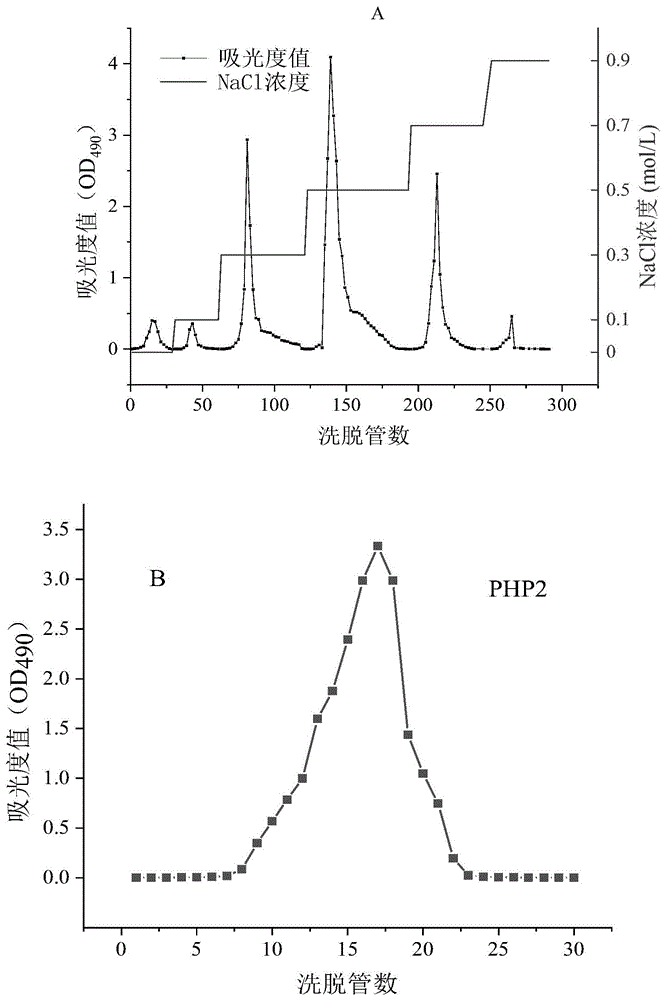
图1是实施例1-1中粗多糖用纤维素deae-52分离的洗脱曲线(a);和0.5mnacl洗脱组分用葡聚糖sephadexg-100凝胶纯化的洗脱曲线(b)。
图2是实施例1-1制备的微胶囊opr形貌图。
图3是实施例1-1制备的微胶囊opr扫描电镜形貌图。
图4是实施例1-1制备的微胶囊opr的ph稳定性(a)、紫外光照射稳定性(b)和贮藏稳定性结果图(c)。
图5是实施例1-1制备的微胶囊opr和对比例1制备的微胶囊or的dpph自由基清除活性结果图。
图6是实施例1-1制备的微胶囊opr的对人宫颈癌细胞hela增殖抑制结果图;
a:hela-24h;b:hela-48h。
图7是实施例1-1制备的微胶囊opr的对人肝癌细胞hepg2增殖抑制结果图;
a:hepg2-24h;b:hepg2-48h。
具体实施方式
实施例1-1、一种制备白藜芦醇的方法(最终溶液中ova与php2质量之比为1:1,ph为3.4时制备的opr微胶囊),依次进行以下步骤:
1)、坛紫菜多糖分离纯化组分的制备(php2的制备):
①、40cm×φ2.6cm的玻璃层析柱中,纤维素deae-52的装填量为100g,每次上样量为400mgcphp(坛紫菜多糖)溶于10ml去离子水。
装配好纤维素deae-52离子交换层析柱并连接恒流蠕动泵和全自动部分收集器,依次以去离子水、0.1m、0.3m、0.5m、0.7m和0.9m的nacl溶液为洗脱剂对cphp(坛紫菜多糖)进行一轮洗脱,控制流速1ml/min,每管10min,用自动收集器收集;当该种洗脱剂没有多糖洗脱下来时换成下一种洗脱剂,可以苯酚-硫酸法追踪每管收集液的吸光度值(od490)作为该种洗脱液洗脱完毕的依据。
合并0.5mnacl溶液洗脱下来的洗脱液,经60℃旋转蒸发浓缩至为原体积的10%,透析脱盐(用截留分子量为3500da的透析袋透析72h)、将所得透析袋中的截留液冷冻干燥36h(真空度≤5pa,-60℃),得初步纯化多糖php-0.5m约250mg。
②、装配好葡聚糖sephadexg-100凝胶层析柱并连接恒流泵与收集器,以去离子水为洗脱液对php-0.5m进行洗脱;具体如下:
玻璃层析柱(50cm×φ1.5)装填12.5g的葡聚糖sephadexg-100,每次洗脱时用100mg的php-0.5m溶于10ml去离子水,去离子水的流速为0.25ml/min,每管20min。用硫酸-苯酚法逐管检测所得洗脱液在490nm处紫外吸收值,绘制sephadexg-100层析柱洗脱曲线(图1b,用于确认收集的管数与样品的纯度,当检测到所得洗脱液不含有多糖时,停止洗脱),收集洗脱曲线上出峰区间内所有试管中的洗脱液,冷冻干燥36h(真空度≤5pa,-60℃),将上述步骤①所得的全部php-0.5m按照进行上样洗脱,共得到纯化多糖php2约80mg。
php2的总糖含量为66.89%,硫酸基含量为8.33%,3,6-内醚半乳糖含量5.6%。单糖组成主要为半乳糖(99.03%),以及少量甘露糖(0.26%)、鼠李糖(0.2%)、葡萄糖(0.1%)等。php2的分子量为523kda。
2)、ova-php2作为壁材制备白藜芦醇微胶囊(opr):
①、分别配制相同浓度(浓度均分别为10g/l)的ova溶液和php2溶液,置于4℃冰箱平衡24h;从而分别得平衡后ova溶液和平衡后php2溶液;
②、将白藜芦醇溶于乙醇,乙醇的量只需能使得白藜芦醇溶解即可,得白藜芦醇乙醇溶液。
③、取由15mg白藜芦醇配制而得的白藜芦醇乙醇溶液加入至500ml的平衡后ova溶液中超声20min(超声参数为100w,40khz),然后用旋转蒸发仪(水浴温度约为40℃)除去乙醇;
再加入500ml平衡好的php2溶液,形成1l复合溶液;该复合溶液中,白藜芦醇的浓度为15mg/l,ova和php2的质量之比为1:1,ova和php2的浓度之和为10g/l;
说明:旋转蒸发除去乙醇时,由于共沸被同时除去的水在整个体系中的含量微乎其微,因此,复合溶液的量仍确定为1l。
将上述复合溶液用磁力搅拌器在60℃下加热搅拌30min,用hcl(1mhcl溶液)调ph至3.4,最后用漩涡振荡器震荡10min,置于4℃冰箱平衡24h。离心(10000rpm,20min),用ph为3.4的hcl溶液清洗沉淀一次(清洗所用的hcl溶液用量浸没过沉淀即可),用真空冷冻干燥机干燥36h(≤5pa的真空度、-60℃的温度),收集的冻干粉即为白藜芦醇微胶囊(opr)。
实施例1-2、最终溶液中ova与php2质量之比为2:1,ph为3.0时制备opr微胶囊:
即,相对于实施例1-1作如下改变:
将步骤2)中的ova溶液和php2溶液的浓度分别改为13.34g/l和6.66g/l;
从而使得复合溶液中,ova和php2的质量之比由“1:1”改成“2:1”;ova和php2的浓度之和保持不变,仍为10g/l,白藜芦醇的浓度仍为15mg/l。
将“调ph至3.4”改成“调ph至3.0”,且,清洗沉淀用的hcl溶液也相应改成ph为3.0的hcl溶液;其余等同于实施例1-1。
实施例1-3、最终溶液中ova与php2质量之比为2:1,ph为3.8时制备opr微胶囊:
即,相对于实施例1-1作如下改变:
将步骤2)中的ova溶液和php2溶液的浓度分别改为13.34g/l和6.66g/l;
从而使得复合溶液中,ova和php2的质量之比由“1:1”改成“2:1”;ova和php2的浓度之和保持不变,仍为10g/l,白藜芦醇的浓度仍为15mg/l。
将“调ph至3.4”改成“调ph至3.8”,且,清洗沉淀用的hcl溶液也相应改成ph为3.8的hcl溶液;其余等同于实施例1-1。
实施例2-1、最终溶液中白藜芦醇浓度为10mg/l,ova和php2在溶液中质量之比为2:1时制得opr微胶囊:
即,相对于实施例1-1作如下改变:
取10mg白藜芦醇配制白藜芦醇乙醇溶液;
ova溶液和php2溶液的浓度分别改为13.34g/l和6.66g/l;
从而使得复合溶液中,ova和php2的质量之比由“1:1”改成“2:1”;ova和php2的浓度之和保持不变,仍为10g/l,白藜芦醇的浓度改为10mg/l。
其余等同于实施例1-1。
实施例2-2、最终溶液中白藜芦醇浓度为5mg/l,ova和php2在溶液中质量之比为3:1时制得的opr微胶囊:
即,相对于实施例1-1作如下改变:
取5mg白藜芦醇配制白藜芦醇乙醇溶液;
ova溶液和php2溶液的浓度分别改为15g/l和5g/l;
从而使得复合溶液中,ova和php2的质量之比由“1:1”改成“3:1”;ova和php2的浓度之和保持不变,仍为10g/l,白藜芦醇的浓度改为5mg/l。
其余等同于实施例1-1。
对比例1:
取消实施例1-1步骤2)中php2的使用,即,取消“加入平衡好的php2溶液”;其余等同于实施例1-1;以此作为空白对照(溶液中,白藜芦醇的浓度为15mg/l),最终得到仅以ova(浓度为10g/l)作为壁材制备的白藜芦醇微胶囊(or)。
实验1、微胶囊中白藜芦醇的包埋率测定
将实施例中2)③步骤中离心后的上清液以及清洗沉淀后的hcl溶液混合并稀释(稀释后的浓度的吸光度值保持在合理范围,即0.2-0.8);307nm下测定吸光度值,离心液下相置于冷冻干燥机冻干(≤5pa的真空度、-60℃的温度)得到复合粒子干粉,然后根据白藜芦醇标准曲线计算上清液中游离白藜芦醇含量,白藜芦醇包埋率和微胶囊载药率计算公式如下:
注:1.白藜芦醇总量即复合溶液中白藜芦醇的总质量,游离的白藜芦醇是指离心后和洗涤沉淀后游离在上清液或者hcl中的白藜芦醇质量,它们的差值就是包埋在微胶囊(即沉淀)中的白藜芦醇质量。
2.opr微胶囊冻干粉质量即实施例中最终冷冻干燥后得到的opr微胶囊冻干粉。
实施例1-1中制备的微胶囊包埋率为95.4%;实施例1-2为85.61%;实施例1-3为85.32%;
实施例2-1为88.91%;实施例2-2为84.92%;对比例1为80.92%。
微胶囊载药率为:实施例1-1为2.89%;实施例1-2为2.45%;实施例1-3为2.19%;
实施例2-1为1.87%;实施例2-2为1.21%;对比例1为2.03%。
实验2、微胶囊复溶液的相关参数表征
a.微胶囊冻干粉复溶液的ζ电位测定:取适量opr干粉复溶于pbs(ph=7,opr浓度为0.05%,w/v,即g/100ml),用激光粒度仪在298k恒温测定复溶液的ζ电位情况。
b.微胶囊冻干粉复溶液的粒径分布以及pdi值:取适量opr干粉复溶于pbs(ph=7,opr浓度为0.05%,w/v),用alv/cgs-3一体式激光散射仪测定不同ph溶液中粒子的粒径分布和多分散性指数(pdi)。
实施例1-1中制备的opr微胶囊,其复溶液的ζ电位为-21.3±1.23mv;粒径为256.8±2.78nm;pdi值为0.4123±0.043。
实验3、微胶囊复溶液的稳定性研究
a.微胶囊冻干粉的ph稳定性研究:分别取适量opr微胶囊冻干粉复溶(用去离子水复溶)使溶液浓度为0.05%(w/v),用hcl或naoh调节至不同ph(2-7),测定不同ph下复合溶液的粒径分布和pdi值。
实施例1-1中的opr微胶囊复溶液随着ph从7降至5,粒径从257.83nm缓慢上升到282.21nm,ph3-4区间,opr微胶囊的粒径开始较快地增大,在ph=3时粒径达到最大值410.49nm;在ph2-7区间内的pdi值均维持在0.4-0.47左右,属于分散度适中的分散体系,而ph=3时的pdi值较ph=7时增大了0.0645,分散液中粒径较为均一,说明opr微胶囊复溶液在中性或弱酸性条件下粒径较小,稳定性良好。
b.微胶囊复溶液的紫外线照射稳定性:分别配制未经包埋的白藜芦醇乙醇溶液(10ml,10mg/l)、or溶液(10ml,浓度0.5mg/ml,去离子水为溶剂)和opr溶液(10ml,浓度0.5mg/ml,去离子水为溶剂),调节ph为7,置于365nm波长紫外光下照射1h,加入适量乙醇(在溶液中浓度≤1%)提取3h,离心后取上清液测定吸光度值并根据标准曲线测定各样品溶液中白藜芦醇含量。
未经包埋的白藜芦醇在紫外光照射下迅速分解,照射20min后白藜芦醇的含量低至50.67%,1h后的白藜芦醇剩余率为48.21%,实施例1-1中的opr中的白藜芦醇20min剩余率为69.28%,1h剩余率为61.01%。说明opr体系有较强的白藜芦醇紫外光分解保护性,降低紫外光照射下的分解速率和分解量。
c.微胶囊复溶液的贮藏稳定性:取将opr复溶后的分散液(0.05%,w/v;ph=7)置于4℃冰箱避光贮藏15d,每隔3d测定其粒径分布和pdi值变化。
实施例1-1中的opr微胶囊在ph=7和4℃避光贮藏15天内的粒径变化不大,粒径从237.41nm增大到261.93nm,pdi值从0.4023增大到0.4389,说明opr微胶囊在中性条件下能保持较小粒径,且粒子粒径分布较均一,opr微胶囊的分散液的贮藏稳定性良好。
其余案例的检测结果如下:
a:ph稳定性研究中,随着ph从7减小到5,实施例1-2的粒径从253.13nm上升到291.93nm;实施例1-3从257.84nm上升到299.38nm;实施例2-1从263.31nm上升到301.32nm;实施例2-2从260.14nm上升到303.38nm;对比例1从258.83nm上升到287.34nm。随着ph继续降低,实施例1-2的最大粒径为439.84nm;实施例1-3为467.45nm;实施例2-1为470.48nm;实施例2-2为471.39nm;对比例1为449.23nm。所有实施例和对比例在ph变化下的pdi值均维持在0.4-0.47左右,属于分散度适中的分散体系。
b.:紫外光照射稳定性实验中,实施例1-2在紫外光照射20min和1h后,微胶囊中白藜芦醇剩余率分别为64.89%和57.31%;实施例1-3分别为60.67%和55.89%;实施例2-1分别为60.56%和57.93%;实施例2-2分别为63.94%和57.99%;对比例1分别为62.89%和53.78%。
c.贮藏14天后,实施例1-2中微胶囊的粒径为271nm;实施例1-3为287nm;实施例2-1为290nm;实施例2-2为256nm;对比例1为250nm;
实验4、微胶囊复溶液的dpph自由基清除活性
配制0.1mmol/ldpph自由基的无水乙醇溶液,取一定质量的or和opr干粉分散到去离子水中,根据载药率分别稀释到分散液中的白藜芦醇的浓度为10、20、30和40μg/ml。以未包埋的白藜芦醇乙醇溶液和维生素c为对照,按照文献(xuj,xull,zhouqw,etal.enhancedinvitroantioxidantactivityofpolysaccharidesfromenteromorphaproliferabyenzymaticdegradation[j].journaloffoodbiochemistry,2016,40(3):275-283.)方法进行操作并计算不同白藜芦醇含量下的opr微胶囊dpph自由基清除率。
未经包埋的白藜芦醇、对比例1中的or以及实施例1-1中的opr在40μg/ml下对dpph清除率分别为55.83%、65.34%和73.48%,ic50值分别为35.77、29.88和24.99μg/ml。
实施例1-2、实施例1-3、实施例2-1、实施例2-2的dpph清除率分别为:69.34%、70.59%、69.34%和65.87%;ic50值分别为26.43、27.45、27.55和26.66μg/ml。
实验5、微胶囊复溶液的抗肿瘤活性
将培养至对数期的hela、hepg2细胞液稀释至合适浓度(即2×104个/ml),吸取100μl细胞液加入到96孔板,每孔细胞数控制在约2000个,在37℃和5%co2环境下贴壁24h,弃孔内细胞培养液并更换100μl新的细胞培养液,然后向每孔加入10μlopr溶液(用去离子水作为溶剂,每孔opr的终浓度0、34.6、173.01、346.02、519.03和692.04μg/ml),空白组加10μlpbs溶液,每种浓度设置5个复孔,轻轻晃动96孔板混匀,继续培养24h或48h,然后按照mtt检测试剂盒说明书准备好相应试剂,向每孔加入10μlmtt溶液,在细胞培养箱内孵育4h,再加入100μl甲瓒溶解液孵育3h,用酶标仪在570nm波长下测定每孔吸光度值。
实施例1-1中opr微胶囊复溶液对hela和hepg2细胞具有显著的抑制活性。剂量为692.04μg/ml时(根据载药率,白藜芦醇为20μg/ml),作用24h下,opr对hela和hepg2细胞的抑制率达30.59和29.76%,抑制两种细胞存活的ic50值为1114.87和1145.59μg/ml;延长作用时间至48h,相同opr剂量下,opr对hela和hepg2细胞48h抑制率有所增加,分别达36.59和39.63%,抑制两种细胞存活的ic50值为927.49和842.06μg/ml。
最后,还需要注意的是,以上列举的仅是本发明的若干个具体实施例。显然,本发明不限于以上实施例,还可以有许多变形。本领域的普通技术人员能从本发明公开的内容直接导出或联想到的所有变形,均应认为是本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!