SU6656在制备治疗疼痛药物方面的应用的制作方法
本发明涉及生物化学领域,具体涉及su6656在制备治疗疼痛药物方面的应用。
背景技术:
src蛋白激酶家族(sfk)包括src、lyn、fyn、lck、hck、fgr、blk、yes和yrk。根据氨基酸序列,sfk可分为2个亚族:一族为c-src、fyn、yes和fgr,在不同组织中广泛表达;另一族为lck、blk、lyn和hck,与造血细胞密切相关。src激酶是目前研究最为深入同时也是与人类疾病联系最为密切的sfk成员,以往的研究发现其通过信号转导调控细胞生长、分化和存活,影响细胞黏附、迁移和侵袭,同时还参与突触传递的调节。
su6656是一src蛋白激酶家族抑制剂。su6656还可以抑制其他蛋白激酶,如camkkα,camkkβ,chk2和srpk1。su6656抑制nih3t3成纤维细胞中pdgf刺激的dna合成和细胞生长,并抑制src和血清促进的细胞增殖。而且,在巨核细胞ut-7/tpo中加入su6656后,细胞停止分离,但是通过核内有丝分裂仍然继续积累dna。su6656还可以减弱放射诱导的akt磷酸化,并提高放射诱导的细胞凋亡和血管内皮细胞损伤。体内实验中,在照射前服用su6656可以显著提高照射诱导的血管损伤,分次照射时服用su6656可以促进肿瘤生长延迟。
su6656(分子式c19h21n3o3s,相对分子质量为371.45,化学结构如式i)主要抑制src,yes,lyn,和fyn。近年来研究发现,su6656可部分阻断高频刺激下c纤维的诱发放电,以及在细胞水平上可抑制钠通道的表达。但至今尚未见到在清醒自由运动的整体动物(invivo)身上观察su6656的镇痛效果的报告。
技术实现要素:
本发明发现了src蛋白激酶家族抑制剂su6656可以明显缓解慢性炎症痛和神经病理性疼痛,从而提供了用于制备治疗抗炎症痛和神经痛药物的应用。
本发明提供的技术方案是:su6656在制备治疗疼痛药物方面的应用。
优选地,su6656在制备治疗抗炎症痛药物方面的应用。
优选地,su6656在制备治疗神经病理性疼痛药物方面的应用。
本发明中的su6656作为主要活性成分制备治疗抗炎症痛药物和疗神经病理性疼痛药物,可以加入一种或多种天然或合成的与其具有协同或辅助作用的活性成分。一种治疗疼痛的药物,其由0.1-100%的su6656和99.9-0%的药用辅料组成药物制剂。
本发明su6656制备的治疗疼痛药物包括药物学上的任何剂型。
本发明中,所述炎症是指外周组织损伤或炎症(如关节或肌肉损伤和炎症)引起的持续性或慢性关节或肌肉痛。
本发明中,所述神经病理性疼痛是指神经损伤引起的持续性或慢性神经痛(如坐骨神经痛,三叉神经痛等)。
剂量和给药途径
本文描述的施用给个体(例如,人类)的su6656剂量可因特定组合物、施用方法、以及病情的特定阶段而变化。其量应足以产生期望的反应。在一些实施方式中,组合物的量是治疗有效量。在一些实施方式中,组合物的量是预防有效量。在一些实施方式中,su6656在组合物中的总量低于诱导毒理学效应的水平(即该水平高于在临床上可接受的毒性水平),或处于当组合物被施用给个体时潜在副作用可被控制或容忍的水平。
在一些实施方式中,su6656在所述药物中的量在以下任一范围内:约0.5mg至约5mg,约5mg至约10mg,约10mg至约15mg,约15mg至约20mg,约20mg至约25mg,约20mg至约50mg,约25mg至约50mg,约50mg至约75mg,约50mg至约100mg,约75mg至约100mg,约100mg至约125mg。
在一些实施方式中,su6656在药物中的给药量至少约为一天0.1mg/kg、0.5mg/kg、1mg/kg、2.5mg/kg、5mg/kg、7.5mg/kg、10mg/kg、15mg/kg或20mg/kg。
药学制剂和给药
根据在本领域中已知的用于制备各种剂型的常规方法,su6656可被配制成溶液、乳液、悬浮液、分散液,或诸如环糊精之类的在合适药物溶剂或载体中的包合配合物,或被配制成丸剂、片剂、锭剂、栓剂、小药囊、糖衣丸、颗粒剂、粉剂、重组粉剂、或连同固体载体的胶囊。实施方式中的药物组合物可通过适当的递送路径被施用,例如口服、肠胃外、直肠、鼻部、局部、或眼部途径,或通过吸入。优选地,组合物被配制成用于静脉内或口服给药。
对于口服给药,药物可以以固体的形式来提供,例如片剂或胶囊,或被配制成溶液、乳液或悬浮液。口服片剂可包括与药学上可接受的相容赋形剂混合的活性组分,所述赋形剂例如稀释剂、崩解剂、结合剂、润滑剂、甜味剂、调味剂、着色剂和防腐剂。适当的惰性填充剂包括碳酸钠和碳酸钙、磷酸钠和磷酸钙、乳糖、淀粉、糖、葡萄糖、甲基纤维素、硬脂酸镁、甘露醇、山梨醇等。示例性的液体口服赋形剂包括乙醇、甘油、水等。示例性崩解剂包括淀粉、聚乙烯基吡咯烷酮(pvp)、淀粉羟乙酸钠、微晶纤维素和藻酸。结合剂可包括淀粉和明胶。润滑剂—如果存在的话—可以是硬脂酸镁、硬脂酸或滑石。如果需要,片剂可通过诸如单硬脂酸甘油酯或二硬脂酸甘油酯之类的材料而被包衣,以延缓在胃肠道中的吸收,或可通过肠溶衣而被包衣。口服制剂可以以下列形式存在:以离散单位的形式,例如胶囊、扁囊剂或片剂,各包含预定量的活性组分;以粉剂或颗粒剂的形式;以溶液或在水性液体或非水性液体中的悬浮液形式;或以水包油液体乳液或油包水液体乳液的形式。活性组分也可被制成大丸剂、药糖剂或糊剂。
口服胶囊包括硬的和软的明胶胶囊。为制备硬明胶胶囊,活性组分可与固体、半固体或液体稀释剂混合。软明胶胶囊的制备可通过将活性组分与以下物质混合而实现:水,诸如花生油或橄榄油之类的油,液体石蜡,短链脂肪酸的甘油单酯与甘油二酯的混合物,聚乙二醇400,或丙二醇。
可通过压制或模制而制成片剂,且可任选一种或多种辅助组分加入到片剂中。可通过在适当的机器中压制以诸如粉末或颗粒之类的自由流动形式存在的活性组分而制得制片剂,且可向其中任选混入粘合剂(例如,聚维酮、明胶、羟丙基甲基纤维素)、润滑剂、惰性稀释剂、防腐剂、分解剂(例如,淀粉羟乙酸钠、交联聚维酮、交联羧甲基纤维素钠)、表面活性剂或分散剂。可通过在适当的机器中对被惰性液体稀释剂润湿的粉状化合物的混合物进行模制而制得模制片剂。片剂可任选被包衣或刻痕,且可被配制成使其中的活性组分缓慢或受控制地被释放,这例如可通过按不同比例使用羟丙基甲基纤维素而实现,以获得所需的释放曲线。
口服液可具有以下形式:悬浮液、溶液、乳液、或糖浆,或可被冻干或呈现为干燥产品,其可在被服用之前使用水或其他适当的载体重新配制。这类液体组合物可任选包含下列物质:药学上可接受的诸如悬浮剂之类的赋形剂(例如,山梨醇、甲基纤维素、藻酸钠、明胶、羟乙基纤维素、羧甲基纤维素、硬脂酸铝凝胶等);诸如油之类的非水性载体(例如,杏仁油或分馏椰子油),丙二醇,乙醇或水;防腐剂(例如,甲基或丙基对羟基苯甲酸或山梨酸);诸如卵磷脂之类的润湿剂;以及—如果需要—调味剂或着色剂。
对于包括静脉内、肌肉内、腹膜内、鼻内或皮下途径在内的肠胃外施用,可将组合物制成如下形式:缓冲至适当ph值和等渗性的无菌水溶液或悬浮液,或肠胃外可接受的油。适当的水性载体包括林格氏溶液和等渗氯化钠。这类形式可被呈现如下:单位剂量形式,例如安瓿或一次性注射装置;多剂量形式,例如从中可抽取适当剂量的小瓶;或可用于制备可注射制剂的固体形式或预浓缩物。适用于包括静脉内给药在内的肠胃外给药的制剂包括以下形式:可包含抗氧化剂的水性和非水性无菌注射液,缓冲液,抑菌剂,以及使制剂与预期接受者的血液等渗的溶质;以及可包含悬浮剂和增稠剂的水性和非水性无菌悬浮液。制剂可被存放在单位剂量或多剂量容器中,例如密封安瓿和小瓶,且可被存储在冷冻干燥(冻干)状态下,仅需在使用之前即刻加入无菌液态载体,例如注射用水。临时注射液和悬浮液可由先前所述的无菌粉剂、颗粒剂和片剂类型制得。
优选的单位剂量制剂是指那些包含一活性组分的每日剂量或单位、每天亚剂量或适当分量的制剂。
附图说明
下面结合附图及实施例对本发明作进一步描述:
图1显示给大鼠腹腔和静脉注射su6656,不影响正常大鼠的机械痛阈和热痛阈。图1a为腹腔注射su6656(n=10)和静脉注射su6656(n=10)前后正常大鼠的机械痛阈。图1b为腹腔注射su6656(n=10)和静脉注射su6656(n=10)前后正常大鼠的热痛阈。
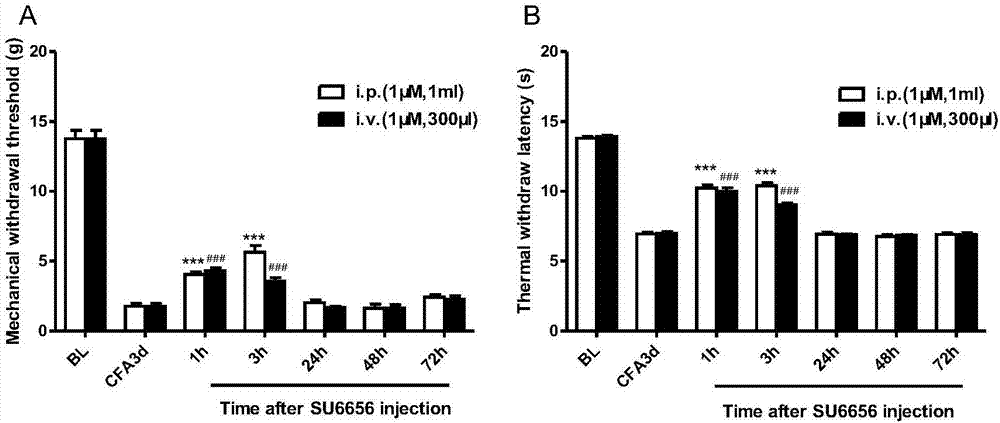
图2显示给大鼠腹腔和静脉注射su6656,可明显翻转cfa大鼠的机械痛敏和热痛敏。图2a为腹腔注射su6656(1μm,1ml)和静脉注射su6656(1μm,300μl)cfa3d大鼠的机械痛阈。图2b为腹腔注射su6656(1μm,1ml)和静脉注射su6656(1μm,300μl)cfa3d大鼠的热痛阈。***p<0.001vs.cfa3d(i.p.);###p<0.001vs.cfa3d(i.v.)。
图3显示给大鼠腹腔和静脉注射su6656,可明显翻转snl大鼠的机械痛敏和热痛敏。图3a为腹腔注射su6656(1μm,1ml)和静脉注射su6656(1μm,300μl)前后snl3d大鼠的机械痛阈。图3b为腹腔注射su6656(1μm,1ml)和静脉注射su66561μm,300μl)前后snl3d大鼠的热痛阈。**p<0.01vs.snl3d(i.p.);##p<0.01vs.snl3d(i.v.);***p<0.001vs.snl3d(i.p.);###p<0.001vssnl3d(i.v.)。
具体实施方式
以下结合具体实施例对上述方案做进一步说明。应理解,这些实施例是用于说明本发明而不限于限制本发明的范围。实施例中采用的实施条件可以根据具体厂家的条件做进一步调整,未注明的实施条件通常为常规实验中的条件。
本实施例运用行为学测定方法,腹腔和静脉注射注射su6656对正常大鼠,炎症大鼠和神经痛大鼠的行为学影响,研究su6656对炎症痛和神经病理性疼痛的镇痛作用。
实验材料
成年雄性健康的sprague-dawley大鼠,体重200~250g,由苏州大学实验动物中心提供。动物中心的卫生级核定文件编号:syxk(苏)2007-0035。实验前,让动物在饲养环境中适应三天。
完全弗氏佐剂购自美国sigma公司。su6656购自美国selleck公司。vonfrey纤维丝购自stoelting公司(nc12775)。足底测痛仪购自iitclifescience公司(model390series8)。
一、动物模型的建立
1.1佐剂性炎症动物模型的建立
采用实验室常规的做法(方法参考bu,f.,etal.,phosphorylationofnr2bnmdasubunitsbyproteinkinasecinarcuatenucleuscontributestoinflammatorypaininrats.scirep,2015.5:p.15945),在正常大鼠右侧后肢足垫皮下注射完全弗氏佐剂(completefreund’sadjuvat,cfa)100μl/只。cfa炎症模型是常用的持续性炎症疼痛模型之一,在注射局部可产生明显炎症现象。选取注射cfa后3天的右侧后肢足垫、踝关节肿胀,表现痛觉过敏的个体作为实验对象即实验组大鼠。
2.坐骨神经分支选择结扎模型(sparednerveligation,snl)动物模型的建立
正常大鼠经4%水合氯醛麻醉后,剪去右侧胫骨部分毛发,碘酊消毒后剪开皮肤,小心分离股二头肌,暴露坐骨神经。分离胫神经、腓总神经和腓肠神经,用5-0的结扎线结扎胫神经和腓总神经,保持腓肠神经完整并不受拉伸及损伤。手术过后涂抹青霉素防止炎症发生。用缝合线缝合肌肉和皮肤。选取snl后3天的右侧后肢足蜷缩,表现痛觉过敏的个体作为实验对象即实验组大鼠。
二、行为学的测定
2.1机械痛阈值的测定(测定方法参考chaplan,s.r.,etal.,quantitativeassessmentoftactileallodyniaintheratpaw.jneuroscimethods,1994.53(1):p.55-63)
大鼠在行为学测定之前,首先要置于铁丝网架上的有机玻璃箱中适应环境,该过程持续三天,每次1h。参照chanplan的经典方法,用vonfrey纤维丝垂直刺激大鼠右后足,刺激的力度以使探针弯成90度为准,持续时间不大于4s,当大鼠出现受试足快速抬起、躲避、舔咬等动作时则视为阳性反应,否则视为阴性反应。当出现阳性反应时给予小一级强度的刺激,当出现阴性反应时给予大一级强度的刺激。当出现一次阳性反应和阴性反应的骑跨,再按上述方法测定4次刺激。最终的机械伤害性缩腿阈(mechanicalwithdrawthreshold,mwt)根据“上下法”计算得到,公式为:50%threshold(g)=(10[xf+kδ])/10000。其中xf为最后一根用于测定的纤维丝的刺激强度,kδ:根据上下法阳性反应和阴性反应出现的模式所对应得到的数值。
2.2热痛阈值的测定(测定方法参考wu,x.b.,etal.,cxcl13/cxcr5enhancessodiumchannelnav1.8currentdensityviap38mapkinaseinprimarysensoryneuronsfollowinginflammatorypain.scirep,2016.6:p.34836.)
大鼠在热痛阈值测定前,首先置于玻璃板上的有机玻璃箱中适应环境,该过程持续三天,每次约10min。在热痛阈测试开始之前,擦拭玻璃板使其保持清洁。使用光源照射大鼠右后足,每组动物在基础热痛阈测定时调整光源至适合强度,并以此强度测定该组动物的后续实验。当大鼠出现缩足,舔咬等行为时,迅速关闭光源,并记下时间值。间隔5min后重复测定,取三次平均值作为热伤害性缩腿阈(thermalwithdrawlatency,twl)。
三、实验及结果和分析
3.1取正常的大鼠20只分成两组,每组n=10,在第一组大鼠的腹腔注射su6656(1μm,1ml),在第二组大鼠静脉注射su6656(1μm,300μl)后,分别经过1h,3h,24h,48h,72h检测各组的机械痛阈和热痛阈。
检测结果如图1所示,显示给大鼠腹腔和静脉注射su6656,不影响正常大鼠的机械痛阈和热痛阈。
3.2取如前述方法制备得到的cfa3d大鼠16只,分成两组,每组n=8,在第一组大鼠的腹腔注射su6656(1μm,1ml),在第二组大鼠静脉注射su6656(1μm,300μl)后,分别经过1h,3h,24h,48h,72h检测各组的机械痛阈和热痛阈。
检测结果如图2所示,显示给cfa3d大鼠腹腔和静脉注射su6656后1h,3h,24h,48h,72h可明显翻转cfa大鼠的机械痛敏和热痛敏。图2a为腹腔注射su6656(1μm,1ml)和静脉注射su6656(1μm,300μl)cfa3d大鼠的机械痛阈。图2b为腹腔注射su6656(1μm,1ml)和静脉注射su6656(1μm,300μl)cfa3d大鼠的热痛阈。***p<0.001vs.cfa3d(i.p.);###p<0.001vs.cfa3d(i.v.)。
3.3取如前述方法制备得到的snl3d大鼠26只,分成两组,每组n=13,在第一组大鼠的腹腔注射su6656(1μm,1ml),在第二组大鼠静脉注射su6656(1μm,300μl)后,分别经过1h,3h,24h,48h,72h检测各组的机械痛阈和热痛阈。
检测结果如图3所示,显示给大鼠腹腔和静脉注射su6656后1h,3h可翻转snl大鼠的机械痛敏和热痛敏。图3a为腹腔注射su6656(n=10)和静脉注射su6656(n=13)前后snl3d大鼠的机械痛阈。图3b为腹腔注射su6656(n=10)和静脉注射su6656(n=13)前后snl3d大鼠的热痛阈。**p<0.01vs.snl3d(i.p.);##p<0.01vs.snl3d(i.v.);***p<0.001vs.snl3d(i.p.);###p<0.001vssnl3d(i.v.)。。
本发明公开的用于制备治疗疼痛药物是由0.1-100%的su6656和99.9-0%的药用辅料组成药物制剂。
为了制备适于胃肠道外途径的溶液剂型,可以使用蒸馏水、注射用水、等渗氯化钠溶液或葡萄糖溶液,或者低浓度(如1-100mm)磷酸缓冲盐水pbs作为载体或赋形剂。、
以上显示和描述了本发明的基本原理、主要特征和本发明的优点。本行业的技术人员应该了解,本发明不受上述实例的限制,上述实例和说明书中描述的只是说明本发明的原理,在不脱离本发明精神和范围的前提下本发明还会有各种变化和改进,这些变化和改进都落入要求保护的本发明范围内。本发明要求保护范围由所附的权利要求书及其等同物界定。
- 还没有人留言评论。精彩留言会获得点赞!