一种pH敏感型透明质酸‑多柔比星纳米前药的制备方法及其应用与流程
本发明涉及生物医药技术领域,具体涉及一种ph敏感型透明质酸-多柔比星纳米前药的制备方法及其应用。
背景技术:
恶性肿瘤是一种严重威胁人类生命的重大疾病,肝癌是常见的消化系统肿瘤之一。多柔比星,又俗称阿霉素、羟柔红霉素,是一种蒽环类广谱抗肿瘤抗生素,周期非特异性药物,对各种生长周期的肿瘤细胞都有杀灭作用。多柔比星能成功抑制多种恶性肿瘤,包括急性白血病、淋巴瘤、软组织和骨肉瘤、儿童恶性肿瘤及成人实体瘤。多柔比星虽然抗瘤谱广、疗效好,但是由于静脉注射后毒副作用较大,主要包括恶心、呕吐、骨髓抑制、胃肠道不良反应和心脏毒性等而严重限制了多柔比星的临床应用。
透明质酸(hyafuronanacid,ha)是一种在细胞外基质、结缔组织和高等动物的器官中广泛分布的天然蛋白多糖。ha是线性单调阴离子聚合物,ha的每个二糖单位均由一分子葡糖醛酸和一分子n-乙酸葡糖胺构成,二糖单位间则由糖普键相连接。天然ha为水溶性高分子,具有多种适宜作为药物载体的优良特性:生物相容性,非免疫原性,在体内可由酶作用而天然降解,并携带大量的羟基和羧基,可进行共价修饰,与药物通过酯化、氢键等结合,达到缓释、控释作用,以这种方式连接的药物,可在体内被细胞摄入或发生降解时释放。
高分子前药是指以高分子化合物为载体的一种药物传递系统,其结构主要由载体、药物、用于连接载体和小分子药物的连接臂以及靶向基团等部分所组成。高分子前药在体内会随着连接基结构的降解,缓慢地释放其共价结合的药物,在抗癌药物的传输领域具有广泛的应用。高分子前药与其他载药系统相比,具有明显的优势:①极大地提高了疏水药物的溶解性,促进药物的吸收;②保护药物在血液循环中的稳定性;③降低药物毒副作用;④改善药物的药代动力学等。
两亲性高分子前药在水介质中可通过自组装形成具有“核-壳”结构的胶束纳米粒,疏水性片段通过疏水相互作用自发聚集,形成纳米粒内核,而亲水性片段则在水溶液中自由伸展,形成纳米粒外壳。一方面,疏水性药物在纳米粒内核中,不仅增加了药物的溶解性,而且降低了药物稳定性,使药物可缓慢释放。与传统的纳米载药系统相比,两亲性高分子前药自组装形成的纳米载药系统具有明显的优势:①在水介质中即可制备纳米颗粒,不需要其他物质的参与,避免了交联剂的毒副作用;②两亲性高分子前药分子直接自组装制备的纳米系统具有较高的热力学稳定性。
技术实现要素:
本发明的目的之一在于提供一种ph敏感型透明质酸-多柔比星纳米前药的制备方法,该方法包括以下步骤:
(a)室温、edc/nhs组合催化剂存在条件下,4-氨甲基苯甲酸甲酯(mepambp)的氨基与高分子载体透明质酸(ha)的羧基发生酰胺化反应,经透析、冷冻、干燥得到产物a(ha-ester),其反应式为:
其中edc为1-乙基-(3-二甲基氨基丙基)碳酰二亚胺,nhs为-羟基琥珀酰亚胺。
(b)在60℃加热回流、氮气保护条件下,产物a的甲氧基与水合肼反应形成联氨,经透析、冷冻、干燥得到产物b(ha-nhnh2),其化学反应式为:
(c)室温、避光条件下,产物b的肼基与多柔比星(dox·hcl)的羰基反应形成具有ph敏感性的腙键,制备得到具有ph敏感性的两亲性高分子前药,其化学反应式为:
(d)具有ph敏感性的两亲性高分子前药采用超声辅助自组装法制备得到高分子前药纳米颗粒。
按照上述方案,步骤(a)中原料ha、edc、nhs、mepambp的投料摩尔比为1:1.27:1.27:1.27。
按照上述方案,步骤(a)具体操作为:将透明质酸溶于甲酰胺中,搅拌条件下分别向溶液中加入edc·hcl和nhs得混合溶液m,将4-氨甲基苯甲酸甲酯溶于甲酰胺中得混合溶液n,将混合溶液n逐滴加入到混合溶液m中,搅拌反应24h后经蒸馏水透析、冷冻、干燥得产物a。
按照上述方案,步骤(b)具体为:将产物a溶于甲酰胺中,再加入过量水合肼,氮气保护下于60℃回流反应16h,经真空除杂、蒸馏水透析、冷冻、干燥后获得产物b。
按照上述方案,步骤(c)具体为:将产物b溶于甲酰胺中,再加入催化剂三乙胺(et3n)和多柔比星,室温下搅拌反应48h后过滤,经真空除杂、蒸馏水透析、冷冻、干燥后获得具有ph敏感性的两亲性高分子前药,其中产物b与多柔比星、三乙胺的摩尔比为2:1:5。
按照上述方案,步骤(d)具体为:将具有ph敏感性的两亲性高分子前药溶于ph=7.4的磷酸盐缓冲液中,得到浓度为5mg/ml的高分子前药溶液,接着对其进行超声处理,超声脉冲宽度5.0s,间歇时间2.0s,超声功率100w,超声处理时间5min。
按照上述方案,所述4-氨甲基苯甲酸甲酯可替换为4-氨甲基苯甲酸乙酯、4-氨乙基苯甲酸甲酯、4-氨乙基苯甲酸乙酯中的一种。
本发明的另一目的是上述ph敏感型透明质酸-多柔比星纳米前药在抗肿瘤药物控制释放和靶向传递领域的应用。
本发明借助于安全无毒、生物相容性好、亲水性强的高分子载体透明质酸,通过腙键使其与抗癌药物多柔比星进行偶联,制备得到了ph敏感型透明质酸-多柔比星纳米前药,该纳米前药在抗肿瘤药物控制释放和抗肿瘤药物靶向传递领域有广阔的应用前景。与现有技术相比,本发明的有益效果如下:
(1)作为高分子载体的透明质酸水溶性、保湿性、生物相容性良好,价廉易得,其可提高多柔比星的溶解性;
(2)该高分子前药纳米粒的自组装过程简单且易于控制,其结构在体内外都具有较高稳定性,易于保存;
(3)该高分子前药具有两亲性,通过自组装形成的纳米粒,可降低多柔比星在体内的生物毒性,提高靶向性和生物利用度以及治疗指数;
(4)本发明所制备的高分子前药纳米粒,其粒径大小在100~120nm,具有epr效应,能够使药物在病灶部位富集,便于定向输送药物。
附图说明
图1为本发明实施例2反应原料、中间产物及终产物的红外光谱图,其中1-a为ha,1-b为ha-ester,1-c为ha-nhnh2,1-d为终产物ha-hyd-dox;
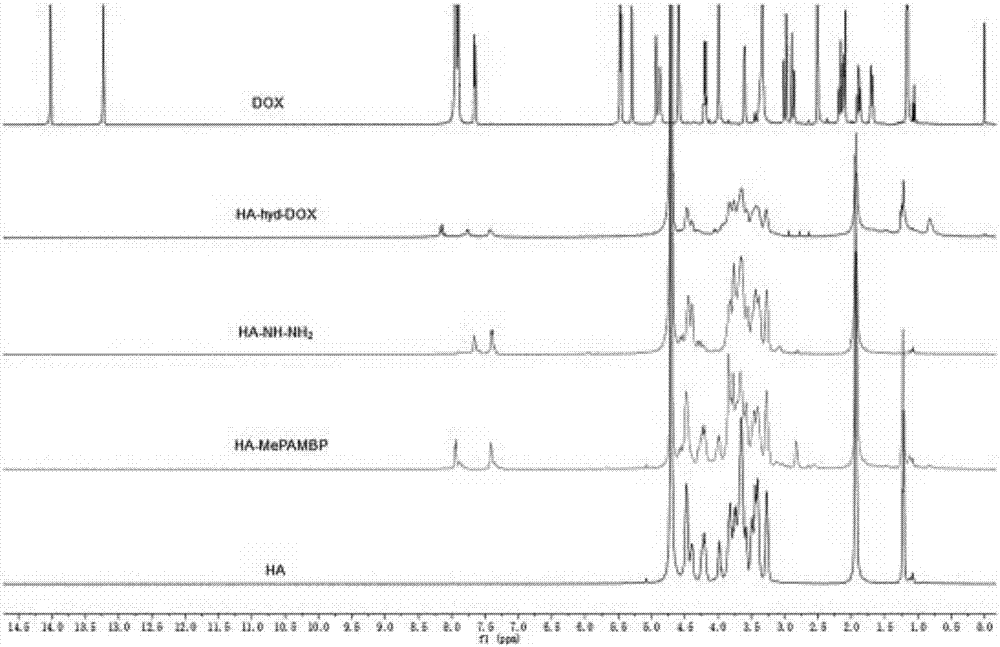
图2为本发明实施例2反应原料、中间产物及终产物的1h-nmr;
图3为本发明实施例2制备的终产物高分子前药纳米颗粒的tem图;
图4为本发明实施例2制备的终产物高分子前药纳米颗粒的粒径分布图;
图5为本发明实施例2制备的终产物高分子前药纳米颗粒在不同ph释放介质中的释药曲线。
具体实施方式
为使本领域普通技术人员充分理解本发明的技术方案和有益效果,以下结合具体实施例进行进一步说明。
实施例1
将400mg透明质酸(ha,分子量为5300da)溶于30ml甲酰胺中,边磁力搅拌边加入256mg1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(edc·hcl),室温条件下反应30min后,加入146mgn-羟基琥珀酰亚胺(nhs),继续反应2h。同时,将255mg4-氨甲基苯甲酸甲酯溶于15ml甲酰胺中,溶解后将其滴加入透明质酸活化体系中,磁力搅拌反应24h。反应结束后,将反应混合物用蒸馏水透析(mwco,3500)3天,冷冻干燥获得产品ha-ester。
称取300mg冻干后的ha-ester溶于15ml甲酰胺中,再加入1.5ml水合肼(nh2-nh2·h2o,质量分数85%),氮气保护条件下于60℃回流反应16h。真空除去大部分溶剂及未反应水合肼后,用蒸馏水透析(mwco,3500)3天,冷冻干燥获得产品ha-nhnh2。
将50mg冻干后的ha-nhnh2溶于10ml甲酰胺,然后加入33μl三乙胺。称取30mg多柔比星(dox·hcl)加入反应液中,室温下磁力搅拌反应48h,过滤。真空除去大部分溶剂后,用蒸馏水透析(mwco,3500)3天,冷冻干燥获得产品ha-hyd-dox。该步反应所有操作均在避光条件下进行。
采用超声辅助自组装法制备纳米颗粒。称取50mg冻干后的ha-hyd-dox高分子前药,溶于一定量的磷酸亚缓冲溶液(pbs,ph=7.4),配制成5mg/ml的高分子前药溶液,然后采用超声波细胞破碎仪超声处理数(超声脉冲宽度5.0s,间歇时间2.0s,超声功率100w,超声处理时间5min,下同),继续用超声清洗器处理几分钟,经过滤膜过滤出去杂质,即可得到ha-hyd-dox纳米溶液。将所制备的ha-hyd-dox纳米溶液冷冻干燥,得到ha-hyd-dox纳米颗粒。
实施例2
将400mg透明质酸(ha,分子量为9800da)溶于30ml甲酰胺中,边磁力搅拌边加入256mg1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(edc·hcl),室温条件下反应30min后,加入146mgn-羟基琥珀酰亚胺(nhs),继续反应2h。同时,将255mg4-氨甲基苯甲酸甲酯溶于15ml甲酰胺中,溶解后将其滴加入透明质酸活化体系中,磁力搅拌反应24h。反应结束后,将反应混合物用蒸馏水透析(mwco,3500)3天,冷冻干燥获得产品ha-ester。
称取300mg冻干后的ha-ester溶于15ml甲酰胺中,再加入1.5ml水合肼(nh2-nh2·h2o,质量分数85%),氮气保护条件下于60℃回流反应16h。真空除去大部分溶剂及未反应水合肼后,用蒸馏水透析(mwco,3500)3天,冷冻干燥获得产品ha-nhnh2。
将50mg冻干后的ha-nhnh2溶于10ml甲酰胺,然后加入33μl三乙胺。称取30mg多柔比星(dox·hcl)加入反应液中,室温下磁力搅拌反应48h,过滤。真空除去大部分溶剂后,用蒸馏水透析(mwco,3500)3天,冷冻干燥获得产品ha-hyd-dox。该步反应所有操作均在避光条件下进行。
采用超声辅助自组装法制备纳米颗粒。称取50mg冻干后的ha-hyd-dox高分子前药,溶于一定量的磷酸亚缓冲溶液(pbs,ph=7.4),配制成5mg/ml的高分子前药溶液,然后采用超声波细胞破碎仪超声处理数分钟,继续用超声清洗器处理几分钟,经过滤膜过滤出去杂质,即可得到ha-hyd-dox纳米溶液。将所制备的ha-hyd-dox纳米溶液冷冻干燥,得到ha-hyd-dox纳米颗粒。
实施例3
将400mg透明质酸(ha,分子量为37000da)溶于30ml甲酰胺中,边磁力搅拌边加入256mg1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(edc·hcl),室温条件下反应30min后,加入146mgn-羟基琥珀酰亚胺(nhs),继续反应2h。同时,将255mg4-氨甲基苯甲酸甲酯溶于15ml甲酰胺中,溶解后将其滴加入透明质酸活化体系中,磁力搅拌反应24h。反应结束后,将反应混合物用蒸馏水透析(mwco,3500)3天,冷冻干燥获得产品ha-ester。
称取300mg冻干后的ha-ester溶于15ml甲酰胺中,再加入1.5ml水合肼(nh2-nh2·h2o,质量分数85%),氮气保护条件下于60℃回流反应16h。真空除去大部分溶剂及未反应水合肼后,用蒸馏水透析(mwco,3500)3天,冷冻干燥获得产品ha-nhnh2。
将50mg冻干后的ha-nhnh2溶于10ml甲酰胺,然后加入33μl三乙胺。称取30mg多柔比星(dox·hcl)加入反应液中,室温下磁力搅拌反应48h,过滤。真空除去大部分溶剂后,用蒸馏水透析(mwco,3500)3天,冷冻干燥获得产品ha-hyd-dox。该步反应所有操作均在避光条件下进行。
采用超声辅助自组装法制备纳米颗粒。称取50mg冻干后的ha-hyd-dox高分子前药,溶于一定量的磷酸亚缓冲溶液(pbs,ph=7.4),配制成5mg/ml的高分子前药溶液,然后采用超声波细胞破碎仪超声处理数分钟,继续用超声清洗器处理几分钟,经过滤膜过滤出去杂质,即可得到ha-hyd-dox纳米溶液。将所制备的ha-hyd-dox纳米溶液冷冻干燥,得到ha-hyd-dox纳米颗粒。
实施例4
将400mg透明质酸(ha,分子量为93000da)溶于30ml甲酰胺中,边磁力搅拌边加入256mg1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(edc·hcl),室温条件下反应30min后,加入146mgn-羟基琥珀酰亚胺(nhs),继续反应2h。同时,将255mg4-氨甲基苯甲酸甲酯溶于15ml甲酰胺中,溶解后将其滴加入透明质酸活化体系中,磁力搅拌反应24h。反应结束后,将反应混合物用蒸馏水透析(mwco,3500)3天,冷冻干燥获得产品ha-ester。
称取300mg冻干后的ha-ester溶于15ml甲酰胺中,再加入1.5ml水合肼(nh2-nh2·h2o,质量分数85%),氮气保护条件下于60℃回流反应16h。真空除去大部分溶剂及未反应水合肼后,用蒸馏水透析(mwco,3500)3天,冷冻干燥获得产品ha-nhnh2。
将50mg冻干后的ha-nhnh2溶于10ml甲酰胺,然后加入33μl三乙胺。称取30mg多柔比星(dox·hcl)加入反应液中,室温下磁力搅拌反应48h,过滤。真空除去大部分溶剂后,用蒸馏水透析(mwco,3500)3天,冷冻干燥获得产品ha-hyd-dox。该步反应所有操作均在避光条件下进行。
采用超声辅助自组装法制备纳米颗粒。称取50mg冻干后的ha-hyd-dox高分子前药,溶于一定量的磷酸亚缓冲溶液(pbs,ph=7.4),配制成5mg/ml的高分子前药溶液,然后采用超声波细胞破碎仪超声处理数分钟,继续用超声清洗器处理几分钟,经过滤膜过滤出去杂质,即可得到ha-hyd-dox纳米溶液。将所制备的ha-hyd-dox纳米溶液冷冻干燥,得到ha-hyd-dox纳米颗粒。
实施例5
将400mg透明质酸(ha,分子量为9800da)溶于30ml甲酰胺中,边磁力搅拌边加入256mg1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(edc·hcl),室温条件下反应30min后,加入146mgn-羟基琥珀酰亚胺(nhs),继续反应2h。同时,将273mg4-氨乙基苯甲酸甲酯溶于15ml甲酰胺中,溶解后将其滴加入透明质酸活化体系中,磁力搅拌反应24h。反应结束后,将反应混合物用蒸馏水透析(mwco,3500)3天,冷冻干燥获得产品ha-ester。
称取300mg冻干后的ha-ester溶于15ml甲酰胺中,再加入1.5ml水合肼(nh2-nh2·h2o,质量分数85%),氮气保护条件下于60℃回流反应16h。真空除去大部分溶剂及未反应水合肼后,用蒸馏水透析(mwco,3500)3天,冷冻干燥获得产品ha-nhnh2。
将50mg冻干后的ha-nhnh2溶于10ml甲酰胺,然后加入33μl三乙胺。称取30mg多柔比星(dox·hcl)加入反应液中,室温下磁力搅拌反应48h,过滤。真空除去大部分溶剂后,用蒸馏水透析(mwco,3500)3天,冷冻干燥获得产品ha-hyd-dox。该步反应所有操作均在避光条件下进行。
采用超声辅助自组装法制备纳米颗粒。称取50mg冻干后的ha-hyd-dox高分子前药,溶于一定量的磷酸亚缓冲溶液(pbs,ph=7.4),配制成5mg/ml的高分子前药溶液,然后采用超声波细胞破碎仪超声处理数分钟,继续用超声清洗器处理几分钟,经过滤膜过滤出去杂质,即可得到ha-hyd-dox纳米溶液。将所制备的ha-hyd-dox纳米溶液冷冻干燥,得到ha-hyd-dox纳米颗粒。
实施例6
将400mg透明质酸(ha,分子量为9800da)溶于30ml甲酰胺中,边磁力搅拌边加入256mg1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(edc·hcl),室温条件下反应30min后,加入146mgn-羟基琥珀酰亚胺(nhs),继续反应2h。同时,将290mg4-氨乙基苯甲酸乙酯溶于15ml甲酰胺中,溶解后将其滴加入透明质酸活化体系中,磁力搅拌反应24h。反应结束后,将反应混合物用蒸馏水透析(mwco,3500)3天,冷冻干燥获得产品ha-ester。
称取300mg冻干后的ha-ester溶于15ml甲酰胺中,再加入1.5ml水合肼(nh2-nh2·h2o,质量分数85%),氮气保护条件下于60℃回流反应16h。真空除去大部分溶剂及未反应水合肼后,用蒸馏水透析(mwco,3500)3天,冷冻干燥获得产品ha-nhnh2。
将50mg冻干后的ha-nhnh2溶于10ml甲酰胺,然后加入33μl三乙胺。称取30mg多柔比星(dox·hcl)加入反应液中,室温下磁力搅拌反应48h,过滤。真空除去大部分溶剂后,用蒸馏水透析(mwco,3500)3天,冷冻干燥获得产品ha-hyd-dox。该步反应所有操作均在避光条件下进行。
采用超声辅助自组装法制备纳米颗粒。称取50mg冻干后的ha-hyd-dox高分子前药,溶于一定量的磷酸亚缓冲溶液(pbs,ph=7.4),配制成5mg/ml的高分子前药溶液,然后采用超声波细胞破碎仪超声处理数分钟,继续用超声清洗器处理几分钟,经过滤膜过滤出去杂质,即可得到ha-hyd-dox纳米溶液。将所制备的ha-hyd-dox纳米溶液冷冻干燥,得到ha-hyd-dox纳米颗粒。
实施例7
将400mg透明质酸(ha,分子量为9800da)溶于30ml甲酰胺中,边磁力搅拌边加入256mg1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(edc·hcl),室温条件下反应30min后,加入146mgn-羟基琥珀酰亚胺(nhs),继续反应2h。同时,将273mg4-氨甲基苯甲酸乙酯溶于15ml甲酰胺中,溶解后将其滴加入透明质酸活化体系中,磁力搅拌反应24h。反应结束后,将反应混合物用蒸馏水透析(mwco,3500)3天,冷冻干燥获得产品ha-ester。
称取300mg冻干后的ha-ester溶于15ml甲酰胺中,再加入1.5ml水合肼(nh2-nh2·h2o,质量分数85%),氮气保护条件下于60℃回流反应16h。真空除去大部分溶剂及未反应水合肼后,用蒸馏水透析(mwco,3500)3天,冷冻干燥获得产品ha-nhnh2。
将50mg冻干后的ha-nhnh2溶于10ml甲酰胺,然后加入33μl三乙胺。称取30mg多柔比星(dox·hcl)加入反应液中,室温下磁力搅拌反应48h,过滤。真空除去大部分溶剂后,用蒸馏水透析(mwco,3500)3天,冷冻干燥获得产品ha-hyd-dox。该步反应所有操作均在避光条件下进行。
采用超声辅助自组装法制备纳米颗粒。称取50mg冻干后的ha-hyd-dox高分子前药,溶于一定量的磷酸亚缓冲溶液(pbs,ph=7.4),配制成5mg/ml的高分子前药溶液,然后采用超声波细胞破碎仪超声处理数分钟,继续用超声清洗器处理几分钟,经过滤膜过滤出去杂质,即可得到ha-hyd-dox纳米溶液。将所制备的ha-hyd-dox纳米溶液冷冻干燥,得到ha-hyd-dox纳米颗粒。
为充分了解本发明制备的ha-hyd-dox高分子前药纳米颗粒的各项性能,分别对其进行了相应的测试,包括ftir、1h-nmr、tem、粒径分布实验以及体外释放性能实验,具体如下:
(1)红外表征
分别对实施例2的原料ha、中间产物ha-ester和ha-nhnh2、终产物ha-hyd-dox取样进行红外光谱分析,所得谱图如图1所示。其中1-a为ha的红外图谱图,3400cm-1处的吸收峰为透明质酸分子上羟基o-h的伸缩振动,在2900cm-1处的吸收峰为亚甲基上c-h的伸缩振动;1650cm-1处的吸收峰为乙酰氨基上c=o的伸缩振动;1560cm-1处的吸收峰为c-n的伸缩振动吸收峰;1400cm-1与1310cm-1处有-c-o-的伸缩振动与-oh的弯曲振动耦合产生的两个吸收峰;1154-944cm-1间有透明质酸糖环的特殊振动吸收峰。以上结果表明透明质酸中存在-cooh、-oh、-ch2和-co-nh-。
1-b为中间产物ha-ester的红外图谱。与1-a相比,在1726cm-1处增加了一个明显而较强的吸收峰,这是ha-ester结构上所新增的甲酯基的吸收峰,说明成功在ha上接枝了酯基。
1-c为ha-nhnh2的红外图谱。与1-b相比,1726cm-1处的吸收峰消失且3500-3400cm-1吸收增强,而且峰值有降低趋势,表明甲酯基团参与了反应,且与水合肼的反应将nh-nh2接枝在ha上,因为3500-3400cm-1吸收增强是由于出现数量较多的-nh2,从而增强了n-h伸缩振动。
1-d为终产物ha-hyd-dox的红外图谱,与1-c相比,在1413cm-1处的吸收峰是dox分子结构上苯环c-c伸缩振动所致,且1618cm-1新出现的吸收峰,为腙键上c=n的伸缩振动所致,从而以此可以表明dox通过腙键成功接枝在了ha上。
(2)核磁共振氢谱的表征
分别对实施例2的原料ha、dox、中间产物ha-ester和ha-nhnh2、终产物ha-hyd-dox取样进行核磁分析,所得谱图如图2所示。与ha相比,ha-ester上的两个化学位移值分别为δ7.45和δ7.40,为对氨甲基苯甲酸甲酯上苯环氢原子的化学位移,说明对氨甲基苯甲酸甲酯成功连接在了ha上。从ha-ester的核磁图谱,可以计算出ha-ester、ha-nhnh2的接枝率:
通过dox与ha-hyd-dox的对比,在δ7.85-8.04上多了两个峰,在dox的谱图上可以详细看出是一个二重峰与一个三重峰,分别为阿霉素蒽环中苯环上对位两个醇羟基中的两个氢和-nh2·hcl上三个氢的峰,因此通过1h-nmr再次证明了ha-hyd-dox被成功合成。
(3)高分子前药纳米粒的表征
实施例2制备得到的终产物ha-hyd-dox高分子前药纳米颗粒的透射电镜图如图3所示,其粒径分布测试结果如图4所示。由图3可知,纳米粒呈规则的球形并均匀分散在水介质。同时,纳米粒在透射电镜测试下的平均粒径约为170nm。结合图4的粒径分析图可知,纳米粒通过动态光散射激光粒度仪测试得到的平均粒径为193.9nm,粒度分布系数为0.106。因此,高分子前药通过自组装的确成功制备了纳米粒。需要提到的是,纳米粒通过透射电镜测试得到的平均粒径要小于通过动态光散射激光粒度仪测试得到的平均粒径,主要是因为透射电镜是在纳米粒干燥的情况下进行的测试,而动态光散射激光粒度仪测试的是纳米溶液,而在溶液的状态下,纳米粒会因为溶剂效应而发生溶胀,因而相比而言其粒径要稍大。
(4)高分子前药纳米粒的体外释放性能研究
以ph=5.0、6.5、7.4的pbs缓冲液来模拟癌细胞内液、癌细胞外基质、人体正常组织体液的ph环境,将它们作为释放介质来考察实施例2制备得到的终产物ha-hyd-dox高分子前药纳米颗粒对ph响应的体外释药行为,结果如图5所示。由图可知,ha-hyd-dox纳米粒在弱酸性环境下的释药速度明显高于纳米粒在生理ph环境中的释药速度。在前12小时之内,ph=7.4和ph=6.5的释放介质中,ha-hyd-dox纳米粒的累积释放百分率分别为11.7%和20.1%,而在ph=5.0的释放介质中纳米粒的累计释放百分率为43.4%。在前48小时之内,ph=7.4和ph=6.5的释放介质中,ha-hyd-dox纳米粒的累积释放百分率分别为13.2%和24.4%,而在ph=5.0的释放介质中纳米粒的累计释放百分率为53.4%。以上结果充分说明了该高分子前药纳米粒具有明显的ph敏感性。
- 还没有人留言评论。精彩留言会获得点赞!