预测肿瘤新生抗原的方法、装置及存储介质与流程
本发明涉及生物信息领域,尤其涉及肿瘤免疫治疗生物标志物发现,具体是指一种对体细胞突变以及基因融合形成肿瘤新生抗原的预测方法及其应用。
背景技术:
肿瘤新生抗原
肿瘤新生抗原是指被人体抗原呈递细胞识别的原本不存于人体的“非我”新生蛋白多肽,该“非我”的新生多肽主要是由肿瘤细胞突变形成的突变蛋白凋亡而来。具体在新生抗原提呈的生物学过程上来说,分为5个步骤:1、抗原呈递细胞(apc)可以通过胞吞肿瘤细胞,将肿瘤细胞内的蛋白(其中包括突变蛋白)裂解成短的肽段;2、apc细胞内的转运蛋白(tap,endosome)将这些肽段转运到内质网上;3、内质网上表达上的hlai类分子,-ⅱ类分子沟槽与肽段锚定结合成稳定的复合物(i类分子结合8~11个氨基酸长度肽段,ⅱ类分子结合13~25个氨基酸长度肽段);4、内质网上的mhc分子与肽段复合物经高尔基体分泌到apc细胞表面;5、免疫t细胞的表面受体tcr识别apc表面的hla分子-肽段复合物,激发后续免疫反应。肿瘤新生抗原是激发机体免疫系统对肿瘤细胞初始免疫反应的关键因素。
肿瘤新生抗原在肿瘤免疫中应用
免疫治疗着眼于恢复机体的免疫系统对肿瘤细胞的识别杀伤能力从而达到祛除肿瘤的目的。不同于对健康细胞与肿瘤细胞通杀的传统放化疗,或者是酪氨酸激酶抑制剂等封闭肿瘤细胞生长生存信号通路的直接杀伤,免疫治疗是一种全新且高效的肿瘤治疗新模式。2013年,美国《科学》杂志将癌症免疫治疗评为“年度十大科学突破”之首。2018年诺贝尔生理学或医学奖也花落免疫治疗领域。尽管免疫治疗发展势头迅猛,然而这一利用免疫系统来攻击肿瘤的策略只对某些癌症及若干病人有效。在没有做任何生物标志物筛选的情况下,绝大部分实体瘤的总缓解率低于30%。而以高微卫星不稳定/错配修复缺失为生物标志物筛选的肿瘤,pd1治疗的总缓解率则可以提高到50%以上。因此,合适的生物标志物来筛选免疫治疗的病人,是实现肿瘤免疫精准医学的关键所在。2018年10月肿瘤突变负荷正式写入美国国立综合癌症网络的非小细胞肺癌诊疗指南,新生抗原作为肿瘤突变负荷触发免疫响应的最终效应因子,可以成为更为准确的生物标志物评估免疫治疗可获益性。以肿瘤新生抗原为基础的肿瘤疫苗个性化治疗也是另一重要应用场景。肿瘤疫苗,是将患者肿瘤细胞中的检测到的新生抗原回输入人体,激发人体免疫反应,定向祛除呈递这些新生抗原的肿瘤细胞。目前,新生抗原,以多肽、核酸或体外经诱导的dc细胞等形式回输人体。ott(pmid:29542692)以及sahin(pmid:28678784),carreno(pmid:25837513)等人,将预测出来的新生抗原,分别以这3种肿瘤疫苗形式应用于在皮肤癌小样本上,取得了很好的治疗效果。综上,肿瘤新生抗原既可作为评估免疫治疗获益的生物标志物,也可以直接应用于肿瘤疫苗的治疗上。
现有的新生抗原预测流程及方法
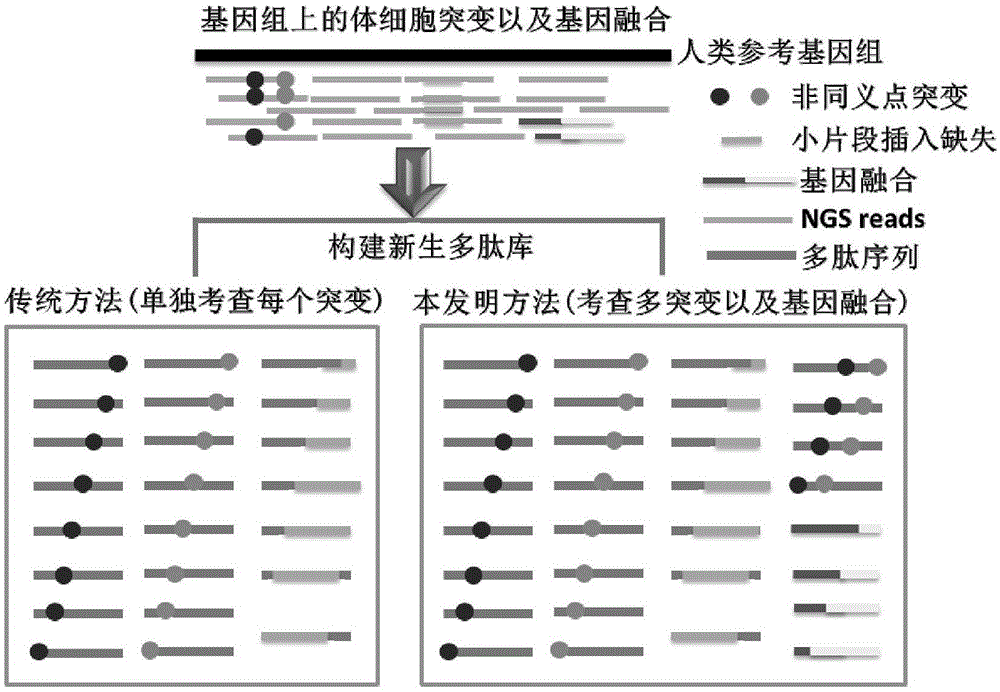
以二代测序技术为基础的全外显子杂交捕获测序,提供了高通量检测肿瘤体细胞突变的可能。目前,新生抗原预测的常见流程是:1、构建新生多肽库,将体细胞突变注释到蛋白水平上,在突变蛋白点周围遍历生成长度为8~11氨基酸、以及13~25氨基酸长度的突变肽以及相应的野生肽段;2、hla分子与新生多肽及其相应野生肽的亲和力预测,基于开源的亲和力预测软件,预测新生多肽以及野生肽与hla分子的亲和力,用经验阈值作过滤,筛选出潜在的新生抗原。通常针对i型与ⅱ型hla分子的亲和力预测软件是丹麦工业大学开发的netmhc,netmhcpan以及netmhcii,netmhciipan(http://www.cbs.dtu.dk/services/)。目前有两大主流开源新生抗原预测软件,分别是西奈山伊坎医学院openvax项目组开发的topiary(https://github.com/openvax/topiary)、以及华盛顿大学麦克唐奈基因组研究所malachigriffith实验室开发的pvactools(https://github.com/griffithlab/pvactools)。这两个开源软件已经应用于多篇发表在权威杂志cancercell,immunity等有关tcga肿瘤大样本数据的研究论文中(pmid:29657128,pmid:29628290)。由于亲和力预测上,topiary以及pvactools均采用netmhc,netmhcpan等工具,这里不做赘述。在新生多肽库生成上,两种软件是以每一个突变为单位生成单突变肽。这里有一个明显的设计缺陷如图1所示,如果在8~11或13~25个氨基酸长度之内发生两个及以上的顺式突变,传统方法会将这些含有多个突变的新生多肽遗失。然而,这些缺失的多点顺式突变形成的新生多肽,也有可能成为机体真正免疫原性的新生抗原。另一方面,这些开源的流程仅仅是把突变形成的突变多肽总称为新生多肽。实际上有一部分突变形成的多肽并非真正的新生多肽,这些突变多肽可能存在于野生型蛋白序列上突变点以外的其它位置上。尤其当这些突变发生在低复杂度序列区域上时(如重复序列上的插入缺失),这种突变多肽不是真正新生多肽的情况更常见。例如,野生型prx蛋白第523~530位的多肽序列为lkvsemkl,第471~478位的多肽序列为pkvsemkl。突变chr19:40902691a>g会导致prx第523位的氨基酸l变为p。此时生成的突变多肽pkvsemkl虽然与523~530位的野生肽序列不一样,但是与野生prx蛋白序列上的471~478位的肽段序列完全一致,因而,此条突变多肽存在于野生型prx蛋白中,对于体机而言并非真正的新生多肽。传统方法的设计缺陷,直接影响了新生抗原预测的准确性。
技术实现要素:
本发明的目的是克服了上述现有技术的缺点,提供了一种更有效的肿瘤治疗响应生物标志物评估、为肿瘤疫苗设计提供精准的候选肽段的预测肿瘤新生抗原的方法。
为了实现上述目的,本发明一方面提供了一种预测肿瘤新生抗原的方法,其具有如下构成:
所述的方法包括步骤:
(1)根据肿瘤-胚系对照样本进行体细胞突变以及基因融合检测;
(2)针对每一对融合基因生成融合肽及相应野生肽;
(3)基于每个体细胞突变生成突变肽及相应野生肽;
(4)构建肿瘤样本特异的个人基因组并生成含有多个突变的多突变肽;
(5)通过突变间的顺式反式关系,判断单突变以及多突变的突变肽真伪,生成真实存在的突变肽;
(6)去除与野生型蛋白其它位置序列完全一致的突变肽,构建完整的新生多肽库;
(7)基于胚系对照样本的bam文件进行hla分子分型检测,预测新生多肽与hla分子的亲和力,将亲和力高的新生多肽作为候选肿瘤新生抗原。
较佳地,所述的步骤(1)的体细胞突变中,在mutect2工具的默认参数下输出体细胞突变结果之后,进行进一步质控过滤,所述的质控过滤包括:突变频率大于2%;突变点的测序深度大于10;至少有2条reads指示有突变且该reads的平均碱基质量>20。
较佳地,所述的步骤(1)中对于基因融合检测,若输入为全外显子wes或全基因因组wgs测序数据,则用factera工具在默认参数下检测基因融合;若输入为rnaseq数据,则用star-fusion工具在默认参数下检测基因融合,然后通过junctionreads数目≥1,做进一步的质控以减少假阳性,所述的junctionreads指直接覆盖融合断点的reads。
较佳地,在所述的步骤(2)中,融合断点注释agfusion根据5’端、3’端断点在基因组上的坐标信息,注释融合断点,并合成融合后的蛋白序列全长;截取长度为l且含有融合断点的融合肽以及对应的5’端、3’端野生肽。
较佳地,具体的截取规则为:
确定5’端融合断点在长度为p5的5’端野生蛋白以及长度为g的融合蛋白上的坐标索引:比对融合蛋白与5’端野生蛋白序列,得出最大的一致性片段序列seq1、以及seq1的5’端野生蛋白上的坐标索引m、融合蛋白上的坐标索引t,所述的seq1的长度为s1,则5’端断点的坐标索引在野生型蛋白上为m+s1,在融合蛋白上为t+s1;
确定3’端融合断点在长度为p3的3’端野生蛋白上的坐标索引:比对融合蛋白与3’端野生蛋白,得出最大一致性片段序列seq2以及seq2在3’端野生蛋白上的坐标索引n,所述的seq2的长度为s2;
截取长度为l的融合肽以及相应5’和3’端野生肽:
在3’端融合断点不造成移码框改变的情况下,每一条融合肽均有相对5’端以及3’端两条野生肽生成,融合蛋白从最小坐标索引t+s1-l到最大坐标索引t,截取长度为l的融合肽;5’端野生蛋白从最小坐标索引m+s1-l到最大坐索引m+s1,3’端野生蛋白从n-l到最大坐标索引n,截取长度为l的两条相应的野生肽;
当3’端融合断点造成移码框改变时,每一条融合肽只有一条5’端野生肽生成,融合蛋白从最小坐标索引t+s1-l到最大坐标索引g-l,截取长度为l的融合肽,5’端野生蛋白从最小坐标索引m+s1-l到最大坐标索引p5-l,顺序生成相应长度为l的野生肽。
较佳地,所述的步骤(3)中,
使用snpeff注释,将每一个体细胞基因组上的碱基突变注释到ensembel数据库上的每一个转录本以及相应蛋白序列上;
截取长度为l的突变肽以及相应的野生肽。
较佳地,截取规则为:
对于错义突变、非移码突变,以突变坐标为中心,向5’端取l-1个氨基酸,向3’端取l-1个氨基酸,生成长度为8~11个氨基酸长度以及13~25个氨基酸长度之间的含有突变氨基酸的突变段以及相应野生肽;
对于典型单点突变,生成38对长度为8~11个氨基酸长度的突变型-野生型肽段,以及247对长度为13~25个氨基酸长度的突变型-野生型肽段;
对于移码突变,从突变点的前l-1个氨基酸开始,延伸到第一个终止子出现为止,生成8~11以及13~25氨基酸长度的突变多段及相应坐标的野生肽段。
较佳地,所述的步骤(4)中,
将体细胞突变的vcf文件中的突变信息一次性全部导入到人类参考基因组上,并替换掉原坐标上的野生型碱基,生成该肿瘤样本所有的个人基因组;
用biopython工具将在基因组碱基水平上编辑过的含有突变的转录本翻译成突变蛋白序列;
基于步骤(3)中单突变注释到的各转录本对应的突变蛋白坐标索引信息,按步骤(3)中的肽段截取规则,生成含有突变的8~11以及13~25氨基酸长度的突变多肽;
所述的步骤(5)中:
通过pysam读取肿瘤样本的bam文件,导出比对到各突变点的reads信息,计算突变点两两之间的
较佳地,所述的步骤(7)中,采用5种不同的二代测序hla分子分型工具,通过生成一致性最高的hla分型结果以减少假阳性;优选地,采用polysolver、hla-hd、hla-prg-la、optitype以及hla-genotyper计算hla分子分型,对8大类hla中的每一个hlaallele,初始得分设为0,每被一个软件检测到,则得分+1,得分最高的hlaallele作为各类hla最终分子分型结果;
预测新生多肽与hla分子的亲和力具体为,当新生多肽与hla分子亲和力<=500nm且相对排位<=2%,则认定为该新生多肽为候选肿瘤新生抗原。
本发明还提供了一种所述的预测肿瘤新生抗原在制备抗肿瘤药物或疫苗的应用。
采用了本发明的预测肿瘤新生抗原及其应用,通过生成肿瘤样本特异的个人基因组,弥补了目前主流方法的两大缺陷:1、对于多突变肽段的错误拆分处理或直接丢失;2、由发生在低复杂度区域突变形成的突变肽段(往往在野生型蛋白中有发现)被误认为新生多肽。从而,使得本发明的新生抗原预测方法能更为全面,准确的反应肿瘤样本真实的新生抗原情况。在13例接受免疫治疗的肝细胞癌数据上,得以证实本发明计算的新生抗原负荷能有效的应用于肿瘤免疫治疗的获益性评估。鉴于本发明在新生抗原上准确全面的预测为肿瘤疫苗设计提供了可靠的肽段来源,肿瘤疫苗上的应用也是本发明另一潜在的应用场景。
附图说明
图1为现有技术方法与本发明在构建新生多肽库上的区别。
图2为本发明提供的新生抗原预测流程示意图。
图3为实施例1中本发明提供的方法与开源软件topiary以及pvactools的比较。
图4为本发明计算在新生抗原在肝细胞癌免疫治疗样本中的生存曲线分析。
具体实施方式
为了能够更清楚地描述本发明的技术内容,下面结合具体实施例来进一步的描述。
本发明提供的一种基于构建肿瘤个人基因组的肿瘤新生抗原预测方法,包括:包含融合断点的融合肽以及相应野生肽截取规则;构建肿瘤个人基因组并生成含有多突变的突变肽;充分考虑肿瘤的异质性特点,利用突变点上ngsreads的jaccard系数衡量突变之间的顺反式关系,保证所生成的突变肽的准确性;基于多个不同工具生成一致性最高的hla分子分型结果,最大程度上保证hla的准确性;去除与野生蛋白中完全一致的突变肽,生成真正意义上的新生多肽。
在本发明提供的方法中:提供了包括来源于基因融合以及体细胞突变的突变肽生成方法,保障突变肽来源的全面性;构建肿瘤样本个人基因组,从而准确生成含有多个突变的突变肽;充分考虑到肿瘤的异质性,运用突变间的顺反式关系,保证了突变肽在机体内的真实情况;去除与野生蛋白完全一致的突变肽,保证了新生多肽的准确性;通过生成多个不同的hla分子分型一致性最高的hla结果,最大程度上提高hla的准确性;对于耗时的hla与新生多肽亲和力预测步骤,进行了并行处理,有效提升了运算效率。
本发明基于全外显子或全基因组测序检测到有体细胞突变,构建肿瘤患者特异的个人基因组,全面考查多点顺式突变的ngsreads信息,提供一种全面准确的新生多肽生成方法。在此基础上,整合主流的亲和力预测方法,计算新生抗原,从而能做更有效的肿瘤治疗响应生物标志物评估,为肿瘤疫苗设计提供精准的候选肽段。
结合图2,说明本发明提供的预测肿瘤新生抗原的方法,该方法包括以下步骤:
step1:针对肿瘤-胚系对照样本对做体细胞突变以及基因融合检测。
开源工具mutect2、factera分别用于体细胞突变以及基因融合检测(对于输入文件为rnaseq数据时,采用star-fusion检测基因融合)。
为保证体细胞突变结果的可靠性,对mutect2默认的参数过滤之后,另做如下质控过滤:
a.突变频率大于2%;b.突变点的测序深度大于10;c.至少有2条reads指示有突变且该reads的平均碱基质量>20。
对于基因融合断点的检测,在factera工具(若为rnaseq数据用star-fusion)的默认参数输出基因融合结果后,另需保证至少一条junctionreads(直接覆盖融合断点的reads)。
step2:针对每一对融合基因生成融合肽及相应野生肽。
用agfusion工具注释融合断点5’端,3’端所在基因的各转录本,并生成相应完整的融合蛋白序列,然后在融合蛋白序列上截取包含融合断点的8-11、13-25氨基酸长度的多肽、以及5’端,3’端对应的野生肽。
具体包括:
1.融合断点注释:agfusion根据5’端,3’端断点在基因组上的坐标信息(具体为染色体号+坐标,如:chr21:42866283),注释融合断点,并合成融合后的蛋白序列全长。
2.截取长度为l含融合断点的融合肽以及对应的5’端,3’端野生肽。
a)确定5’端融合断点在长度为p5的5’端野生蛋白以及长度为g的融合蛋白上的坐标索引。比对融合蛋白与5’端野生蛋白序列,得出最大的长度为s1的一致性片段序列seq1,以及seq1在5’端野生蛋白上的坐标索引m,在融合蛋白上的坐标索引t,则5’端断点的坐标索引在野生型蛋白上为m+s1,在融合蛋白上为t+s1。
b)确定3’端融合断点在长度为p3的3’端野生蛋白上坐标索引。比对融合蛋白与3’端野生蛋白,得出长度为s2的最大一致性片段序列seq2,以及seq2在3’端野生蛋白上的坐标索引n。
c)截取长度为l的融合肽以及相应5’和3’端野生肽。
在3’端融合断点不造成移码框改变的情况下,每一条融合肽均有相应5’端以及3’端两条野生肽生成。融合蛋白从最小坐标索引t+s1-l到最大坐标索引t,截取长度为l的融合肽。5’端野生蛋白从最小坐标索引m+s1-l到最大坐索引m+s1。3’端野生蛋白从n-l到最大坐标索引n截取长度为l的两条相应的野生肽。
当3’端融合断点造成移码框改变时,每一条融合肽只有一条5’端野生肽生成。融合蛋白从最小坐标索引t+s1-l到最大坐标索引g-l,截取长度为l的融合肽。5’端野生蛋白从最小坐标索引m+s1-l到最大坐标索引p5-l,开始截取长度为l的野生肽。
step3:基于每个体细胞突变生成单突变肽及相应野生肽。
用snpeff注释将基因组上的体细胞碱基突变注释到到ensembel数据库上的每一个转录本以及相应蛋白序列上。截取包含有蛋白突变位点长度为8-11、13-25氨基酸长度的突变肽、以及相应位置上的野生肽。
具体包括:
a)用snpeff注释将每一个体细胞基因组上的碱基突变注释到ensembel数据库上的每一个转录本以及相应蛋白序列上。
b)截取长度为l的突变肽以及相应的野生肽。
对于错义突变,非移码突变而言,以突变坐标为中心,向5’端取l-1个氨基酸,向3’端取l-1个氨基酸(l为所要生成的突变肽长度)。生成长度为8-11个氨基酸长度以及13-25个氨基酸长度之间的含有突变氨基酸的突变段以及相应野生肽。
对于一个典型的单点突变而言,可以生成38对长度为8-11个氨基酸长度的突变型-野生型肽段,以及247对长度为13-25个氨基酸长度的突变型-野生型肽段。
如若突变为移码突变,则从突变点的前取l-1个氨基酸开始,延到伸到第一个终止子出现为止,生成8-11以及13-25氨基酸长度的突变多段及相应坐标的野生肽段。
step4:构建肿瘤样本特异的个人基因组并生成含有多个突变的突变肽。
批量将step1检测到的所有突变碱基,替换掉人类参考基因组上的碱基。此优点在于,可以同时捕捉到每个基因上出现的多个突变。
对包含多个突变的突变肽的遗失或错误注释,是现有新生抗原预测工具的一大短板。本发明采用生成肿瘤样本特异的个人基因组的方法,用以弥补这一缺陷。具体地,将体细胞突变的vcf文件中的突变信息一次性全部导入到人类参考基因组,并替换掉原坐标上的野生型碱基,生成该肿瘤样本所有的个人基因组。
用biopython工具将在基因组碱基水平上编辑过的含有突变的转录本翻译成突变蛋白序列。基于step3单突变注释到的各转录本对应的突变蛋白坐标索引信息,按step3中的肽段截取规则,生成含有突变的8-11以及13-25氨基酸长度的突变多肽。
相较于基于单突变注释信息在蛋白水平上编辑,本发明通过编辑基因组的碱基生成肿瘤个人基因组,然后统一注释整个转录本,能够更为准确的反应多个碱基突变之后的突变蛋白情况。尤其当点突变发生在同一个氨基酸的三联密码子之内时。如:chr11:56143803a>g,chr11:56143804g>a发生在同一个密码子内,单独注释到蛋白水平时,分别为orbu1:p.gln235arg,以及orbu1:p.gln235his,会造成蛋白水平编辑的冲突。此步骤需要注意保证突变对应的参考基因组版本与将导入的参考基因组版本一致。目前此发明支持人类参考基因组的主流版本grch37和grch38,可以进一步扩展到其它物种的参考基因组上如大鼠、小鼠等。
step5:通过突变间的顺式反式关系判断step3中生成的单突变以及step4中生成的多突变肽真伪,生成真实存在的突变肽。
根据序列比对bam文件中的reads信息,确定突变间的关系是顺式还是反式关系,用以判断含有多突变以及单突变的突变肽真伪。以两个突变为例,如果两个突变为反式突变,即没有一条reads是同时包含这两个突变,则将这将含有该双突肽的突变肽去除,仅保留含单个突变的突变肽。如果两个突变为顺式突变,则仅保留同时含这两个突变的突变肽。
由于肿瘤异质性,体细胞突变文件中的所列突变并非全部是顺式突变,即这些突变不一定是出现在同一个肿瘤亚克隆中,或者说多个突变是分散在不同的亚克隆中,在测序数据上表现为多个突变出现在不同的ngsreads上。因此,此步骤中引入肿瘤样本序列比对的bam文件用于评判这些多突变肽段以及单突变肽段的真伪。只有指示这几个突变的ngsreads有部分重叠,才能保证step4生成的含有多突变肽以及step3生成的单突变肽段都是真实存在的。
具体是通过pysam读取肿瘤样本的bam文件,导出通过各突变点的染色体坐标的reads信息,计算突变点两两之间的
step6:去除与野生型蛋白其它位置序列完全一致的突变肽,构建完整的新生多肽库。
新生多肽是指突变引起的不存在于野生型蛋白上突变肽,这样才会被机体免疫系统认为是新生的。经由step5形成的突变肽并非完全等同于新生多肽。尤其像发生的重复区域的蛋白突变,很容易在野生型蛋白的其它位置找到与突变肽完全一致的序列,这种肽段并不能称为真正的新生抗肽段。这点也是现有开源工具直接忽略掉的地方。此步骤针对每条突变肽,通过pyensemble获取每个转录本对应的野生蛋白质序列,检查突变肽是否在野生蛋白质序列中出现。
step7:基于胚系对照样本的bam文件做高一致性高的hla分子分型检测。
考虑到作为hla分子分型检测的金标准一代测序一致性也仅达84%(pmid:27802932),本发明采用5种不同的二代测序hla分子分型工具,通过生成一致性最高的hla分型结果以减少假阳性。具体地,用polysolver、hla-hd、hla-prg-la、optitype以及hla-genotyper计算hla分子分型。其中polysolver、optitype仅计算i型hla检测,其它三种同时也可用于ⅱ型hla检测。对8大类hla(a,b,c,drb,dpa,dpb,dqa,dqb)中的每一个hlaallele,初始得分设为0,每被一个软件检测到,则得分+1。得分最高的hlaallele作为各类hla最终分子分型结果。
step8:预测新生多肽与hla分子的亲和力,并根据亲和力高低,计算样本的新生抗原负荷。
新生抗原的预测实际上模拟抗原呈递细胞中的hla分子通过结构上的凹槽与新生多肽的锚定结合。现有几个主流工具如netmhc、netmhcpan、netmhcii、netmhciipan、mhcflurry等均是基于对真实数据中hla分子与短肽的亲和力来训练每个hla分子特异的神经网络模型,然后用于该hla分子与新生多肽的亲和力预测。目前这类算法几乎被所有已报道的新生抗原预测工具所应用。本发明也采用这几种主流的亲和力预测算法。
具体地筛选条件为,当新生多肽与hla分子亲和力<=500nm且相对亲和力排位<=2%,则认定为该新生多肽为新生抗原。这里面输出的亲和力用ic50值表示,代表与50%的hla分子结合时的该新生多肽浓度,单位为nm。该数值越小,表示肽段与该等位基因所编码的hla蛋白的亲和力越高。亲和力相对排位用rank(%)表示。即该新生多肽的ic50值在随机生成的400000条肽段的ic50数据集中的相对百分排位。数值越小,说明肽段与该hla分子的亲和力处于相对越高的位置。达到这一阈值的所有新生抗原-hla分子复合物的总数称之为肿瘤新生抗原负荷。
实施例1
本实施例是以一例非小细胞肺癌样本的突变文件开始,具体突变信息见表1,且分别比较topiary、pvactools以及本发明的方法预测8-11个氨基酸长度的新生多肽。
该实施例可以为本发明的验证方案实施实例,证明本发明相较于目前主流的两个工具的优势。图3说明了本发明的新生抗原预测方法与主流的两个开源工具的比较过程与结果。由于三种工具对于肽段与hla分子的亲和力采用相同的方法,这里只关注三种工具在新生多肽生成上的差异。
表1非小细胞肺癌患者的58个体细胞突变位点信息
本发明提供的方法中,根据在野生蛋白中是否检测到作为判断生成的新生多肽真伪的依据,突变而形成的所有突变多肽会成两部分:真的新生多肽、假的新生多肽。在此例中,本发明共生成203条假的新生多肽(即突变多肽序列能在野生型蛋白中有发现),1792条真的新生多肽。topiary与pvactools分别生成1748条、1702条新生多肽。
从三个工具的结果比较中,可以明显看到3点发现:1、本发明与pvactools方法生成的新生多肽能完全覆盖topiary的结果;2、topiary与pvactools预测的新生多肽序列中有62条在野生型蛋白中发现。这种笼统的将突变而来的突变多肽称之为新生多肽的做法有欠考虑;3、pvactools与topiary在处理相邻的双碱基突变时有欠缺。对于双碱基替换突变,pvactools直接强行拆分为两个碱基突变,会造成氨基酸注释错误,topiary则直接忽略掉全部双碱基突变(此样本中共4个)导致生成的新生多肽减少。pvacseq所特有76条错误的新生多肽来自于4个突变:chr15:28947425g>a;chr15:28947426a>g;chr4:145041707c>a;chr4:145041708t>c。该4个突变是由直接将体细胞突变文件中的两个相邻双碱基突变chr15:28947425:ga>ag;chr4:145041707ct>ac强行拆分而来。仔细分析本发明所特有的190条真的新生多肽,发现主要集中在表2所示的9个突变上。其中有4个双碱基突变丢失,尤其缺失一个egfr热点突变形成的新生多肽。此外,还有3个突变丢失了部分转录本的注释,而导致相应新生多肽的遗失。
综上说明,本发明达到了预期的设计效果,能弥补现有工具缺陷,这将有助于精确计算肿瘤新生抗原负荷并评估免疫治疗效果,以及提供可靠的多肽信息服务于后期的肿瘤疫苗设计。
表2本发明特有的190条新生多肽所对应的9个突变信息
实施例2
实施例2为本发明提供的方法在肿瘤免疫治疗上具体应用场景,用以说明本发明在肿瘤免疫治疗上的应用价值,以及相较于目前已获批的肿瘤突变负荷的优势。高肿瘤突变负荷说明有更多的肿瘤体细胞突变,意味着能产生更多的肿瘤新生抗原,这样肿瘤细胞被免疫细胞识别的可能性也就越大,这正是肿瘤突变负荷作为生物标志物评估免疫治疗效果的生物学理论所在。实施例2验证本发明计算的肿瘤新生抗原负荷作为免疫治疗生物标志物的有效性。
表3中列有经免疫治疗的13例肝细胞癌病人的总体生存数据,以及经过全外显子测序检测到的肿瘤突变负荷,本发明计算的样本新生抗原负荷。在此处,肿瘤突变负荷(tmb)定义为全显子上检测到的非同义体细胞突变个数。肿瘤新生抗原负荷(tnb)是指所有满足阈值(新生多肽与hla分子亲和力<=500nm且相对排位<=2%)的新生多肽-hla分子复合物的总数。根据tmb的中位值,可将病人分为两组:高tmb组7人,低tmb组6人。相同的,也可以根据tnb的中位值,将病人分为高tnb组以及低tnb组。在图4中,分别作了tnb、tmb高低分组的生存曲线。发现tnb能显著区分免疫治疗病人的生存情况(p值<0.05,高tnb组osvs低tnb组os为565天:185天)。虽然在趋势上可以看到高tmb组相比低tmb组有延长的os,统计学上不具有显著性(p值=0.29,高tmb组osvs低tmb组os为336天:304天)。此结果表明,本发明的新生抗原预测方法相比肿瘤突变负荷能更为精确的评估肿瘤免疫治疗的效果。新生抗原负荷作为生物标志物有着很好的应用场景。
表3
综上,本发明的新生抗原预测方法在新生多肽生成上,通过生成肿瘤样本特异的个人基因组,弥补了目前主流方法的两大缺陷:1、对于多突变肽段的错误拆分处理或直接丢失;2、由发生在低复杂度区域突变形成的突变肽段(往往在野生型蛋白中有发现)被误认为新生多肽。此外,本发明也同时纳入体细胞突变以及基因融合形成的新生多肽。从而,使得本发明的新生抗原预测方法能更为全面,准确的反应肿瘤样本真实的新生抗原情况。在13例接受免疫治疗的肝细胞癌数据上,得以证实本发明计算的新生抗原负荷能有效的应用于肿瘤免疫治疗的获益性评估。鉴于本发明在新生抗原上准确全面的预测为肿瘤疫苗设计提供了可靠的肽段来源,肿瘤疫苗上的应用也是本发明另一潜在的应用场景。
在此说明书中,本发明已经参照其特定的实施例作了描述。但是,很显然仍可以作出各种修改和变换而不背离本发明的精神和范围。因此,说明书和附图应被认为是说明性的而非限制性的。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种具抗肿瘤活性的不对称双1‑取代5‑N杂靛红席夫碱类化合物的合成方法与制造工艺
- 一种具抗肿瘤活性的不对称双7‑N杂靛红席夫碱类化合物的合成方法与制造工艺
- 一种具抗肿瘤活性的不对称双取代靛红席夫碱类化合物及其合成方法与制造工艺
- 一种全人源抗MAGEA1的全分子IgG抗体及其应用
- 基于特异性单克隆或多克隆抗体建立的ny-eso-1抗原检测方法
- 一种规模化制备人前列腺癌pc-3细胞的方法
- 一种肿瘤抗原gpc3的ctl识别表位肽及其应用
- 一种<sup>18</sup>F标记的多肽化合物及其制备方法和应用
- 一种靶向Mesothelin的复制缺陷性重组慢病毒CAR-T转基因载体及其构建方法和应用
- 一种靶向cd123的复制缺陷性重组慢病毒car-t转基因载体及其构建方法和应用