一种苯甲酰胺小分子抑制剂及在抑制鸟氨酸脱羧酶上的应用的制作方法
本发明涉及鸟氨酸脱羧酶(ornithinedecarboxylase;odc)的小分子抑制剂及其应用,具体涉及人源鸟氨酸脱羧酶的小分子抑制剂及其在抑制和杀伤肿瘤细胞中的应用。
背景技术:
蛋白质是生物体的主要组成成分之一,是完成各种生命活动的主要物质。在各种蛋白质中,蛋白酶对生命活动至关重要,几乎所有生物体内的生化反应过程都由蛋白酶进行催化。生物体内各种蛋白酶的活性具有严格的调控机制,一旦其调控机制出现问题,造成蛋白酶的活性过高、过低或者是完全失活,都会造成相应的各类疾病。因此,通过药物来调控蛋白酶的活性,使其恢复和保持在正常水平,具有非常重要的理论意义和现实意义。以结构为基础的药物设计,是设计以蛋白质为靶标的药物的一个非常重要的手段。
多胺(polyamines)是一类从氨基酸代谢产生的带正电的阳离子小分子,在所有生物体中都存在,对细胞生长、分化、存活及正常生物学功能等都不可缺少。多胺带多正电荷的特性,使它们能通过与带负电荷的生物大分子(dna、rna、蛋白质、细胞膜等)形成静电作用,从而调控非常广泛的生物学过程,包括染色体结构形成、dna合成与稳定、dna复制、转录和翻译、蛋白质磷酸化、核糖体生成、离子通道和膜表面受体的调控、自由基清除等。天然的多胺有很多种。在哺乳动物中,天然存在的有三种,即腐胺(putrescine),精脒(spermidine),精胺(spermine),它们对哺乳动物正常生长和发育必不可少。由于多胺具有重要的生物学功能,其细胞内水平受到严格的调控。在快速繁殖的细胞中,如肿瘤细胞,多胺水平及odc表达水平也会上升和失调。多胺水平升高,伴随着细胞增殖加快、凋亡减少、以及肿瘤浸润和转移相关基因的表达水平升高等。因此,多胺的调控,成为肿瘤治疗和药物研发中的一个重要手段。
多胺代谢的起始底物是鸟氨酸(ornithine),它是精氨酸在尿素循环(ureacycle)中被精氨酸酶(arginase)催化的反应产物。odc是多胺合成途径的第一个酶,催化从鸟氨酸(ornithine)到腐胺的反应,该步催化反应也是多胺合成途径的一个限速步骤。因此,合成odc抑制剂,抑制腐胺的生成,是目前很受关注的一个肿瘤治疗途径。同时,由于病原微生物也需要正常的多胺水平,odc抑制剂也成为针对病原微生物(如导致非洲锥虫病的布氏锥虫)的重要靶标。
当前,odc的抑制剂dfmo(α-二氟甲基鸟氨酸)已经被用于临床,辅助癌症的化疗。但其与odc的结合能力较弱,作用浓度很高,且由于是与odc形成共价键的自杀性抑制剂,毒副作用非常大。因此,非常有必要开发出具有更好效果的odc新型抑制剂。
技术实现要素:
本发明的目的是通过计算机辅助的高通量药物筛选,提供一种针对odc的新型小分子抑制剂,将其应用于鸟氨酸脱羧酶的抑制剂,可能可以用于制备治疗肿瘤、病原微生物感染的药物。具体为:
一种苯甲酰胺小分子抑制剂,该小分子抑制剂的结构式为:
采用上述苯甲酰胺小分子抑制剂在抑制鸟氨酸脱羧酶上的应用。
采用苯甲酰胺小分子抑制剂在制备治疗肿瘤药物中的应用。
将上述苯甲酰胺小分子抑制剂用于抑制鸟氨酸脱羧酶的方法,包括如下步骤:
1)odc原核表达质粒的构建
将odc的基因序列,通过bamhi和xhoi酶切位点,插入到pet28a质粒中,构建出pet28a-hodc质粒,经dna测序验证;
2)odc蛋白的表达
将步骤1)构建的pet28a-hodc质粒通过cacl2法,转化到大肠杆菌bl21菌株中,通过卡那霉素进行筛选,然后将在含有卡那霉素的luria-bertani(lb)培养板上生长的菌株,接种至含有卡那霉素的lb液体培养基中,在37℃、250rpm培养至对数生长期,再添加异丙基硫代半乳糖苷(iptg)至0.5mm,在28℃诱导表达4小时,最后,离心收集细菌;
3)odc蛋白的纯化
将步骤2)中收集的细菌,用裂解液重新悬浮,然后通过超声方法进行细胞裂解,再将裂解液在4℃、12000转/min,离心后,保留上清;最后将上清利用ni-ntahis标签蛋白结合填料进行结合和纯化,得到人源odc蛋白,odc蛋白的洗脱缓冲液为50mm三羟甲基氨基甲烷(tris)/hcl,ph8.0,300mmnacl,1mmdtt,100mm咪唑(imidazole);
4)odc蛋白的活性检测
在ep管中,加入400ul底物反应混合物、50ug的odc蛋白,混匀后将ep管放置在37℃水浴中30min;加入400ul10%的三氯乙酸终止反应,室温离心5000rpm、5min,然后取出100ul上清,与200ul4mol/l的naoh溶液混合,加入400ul正戊醇,充分混合,2000rpm离心5min,再将上层液200ul转移至新的ep管中,加入200ul0.1mol/lph为8.0的四硼酸钠混合均匀、加入200ul10mmol/l三硝基苯磺酸混合充分、加入400uldmso,充分混合1min,3000rpm离心5min;最后取出上层液到96孔板中,用酶标仪检测426nm处的吸光值,得到未加酶的od值.
5)抑制剂对odc蛋白的抑制活性的检测
根据步骤4)中所述的方法,在加入400ul底物反应混合物后,随即加入鸟氨酸脱羧酶的小分子抑制剂,后续操作同步骤4);
通过以下公式计算出odc抑制率:
对照差=加小分子抑制剂对照组平均od值-未加小分子抑制剂对照组平均od值,其中对照组在步骤5)中加入的抑制剂为dfmo抑制剂;
实验差=加小分子抑制剂实验组平均od值-未加小分子抑制剂实验组组平均od值;
odc抑制率=[(对照差-实验差)/对照差]×100%。
所述的步骤3)中所述的裂解液为50mm三羟甲基氨基甲烷(tris)/hcl,ph8.0,300mmnacl,1mmdtt,1mmpmsf,5mm咪唑(imidazole)的混合液。
所述的步骤4)中的底物反应混合物为在150mmph为7.1的磷酸盐缓冲液(pbs)中溶解17.57ulβ-巯基乙醇,55.84mg1.5mmedta二盐钠,75nm磷酸吡哆醛(plp)储液,2mm鸟氨酸盐酸盐。
所述的鸟氨酸脱羧酶为人源鸟氨酸脱羧酶、非人源鸟氨酸脱羧酶或与人源鸟氨酸脱羧酶的腐胺底物及磷酸吡哆醛结合位点高度同源的蛋白质。
按照以上方案,首先利用pocket蛋白质药物口袋分析软件,以人源odc的晶体结构为基础,对其腐胺底物及plp配体结合口袋进行分析,并产生一个该蛋白质口袋的理论模型。然后,利用蛋白质对接程序dock,将specs小分子药物库中的19万个小分子,对接到上述蛋白质口袋模型中,并筛选出至少含15个非氢重原子、至少形成2个氢键、至少1个疏水中心、对接打分不超过-10的小分子。再对上述小分子进一步利用蛋白质-小分子对接程序autodock,再一次依次对接到上述蛋白质口袋中,进行进一步的对接计算,最后挑选出对接打分低于-7的小分子。
本发明还提供一种组合物,其含有对抑制鸟氨酸脱羧酶有效量的本发明提供的小分子抑制剂或其类似物和药学上适用的载体。优选所述组合物是药物组合物,其含有对治疗有效量的本发明提供的小分子抑制剂或其类似物和药学上适用的载体。更优选所述药物组合物是治疗或预防鸟氨酸脱羧酶(odc)的抑制产生应答的疾病的药物组合物,其中鸟氨酸脱羧酶(odc)优选人源性鸟氨酸脱羧酶(odc);还包括含有对于防治鸟氨酸脱羧酶的抑制产生应答的疾病有效量的本发明提供的小分子抑制剂或其类似物以及药学上适用的载体。
本发明的小分子抑制剂或其类似物以及上述组合物可用于抑制鸟氨酸脱羧酶(odc),优选人源性鸟氨酸脱羧酶(odc),其中所述抑制是治疗目的或非治疗目的。优选本发明的小分子抑制剂或其类似物用于制备抑制鸟氨酸脱羧酶(odc)的药物,优选人源性鸟氨酸脱羧酶(odc)。因此本发明还提供了抑制鸟氨酸脱羧酶活性的方法,该方法包括给需要抑制鸟氨酸脱羧酶(odc)的目标物施用有效量的本发明的小分子抑制剂或其类似物或上述组合物,其中所述抑制是治疗目的或非治疗目的。
本发明还提供了治疗对鸟氨酸脱羧酶的抑制产生应答的疾病的方法,该方法包括给需要这种治疗或预防的个体施用预防或治疗上有效量的抑制鸟氨酸脱羧酶(优选人源性鸟氨酸脱羧酶(odc))的本发明的小分子抑制剂或其类似物或上述组合物。
本发明的小分子抑制剂或其类似物可用于制备治疗对鸟氨酸脱羧酶的抑制产生应答的疾病的药物或药物组合物,其中鸟氨酸脱羧酶(odc)或其类似物抑制鸟氨酸脱羧酶的活性,所述疾病包括肿瘤或病原微生物感染,优选上述肿瘤。原虫感染是指对鸟氨酸脱羧酶的抑制产生应答的肿瘤疾病或病原微生物感染疾病。
附图说明
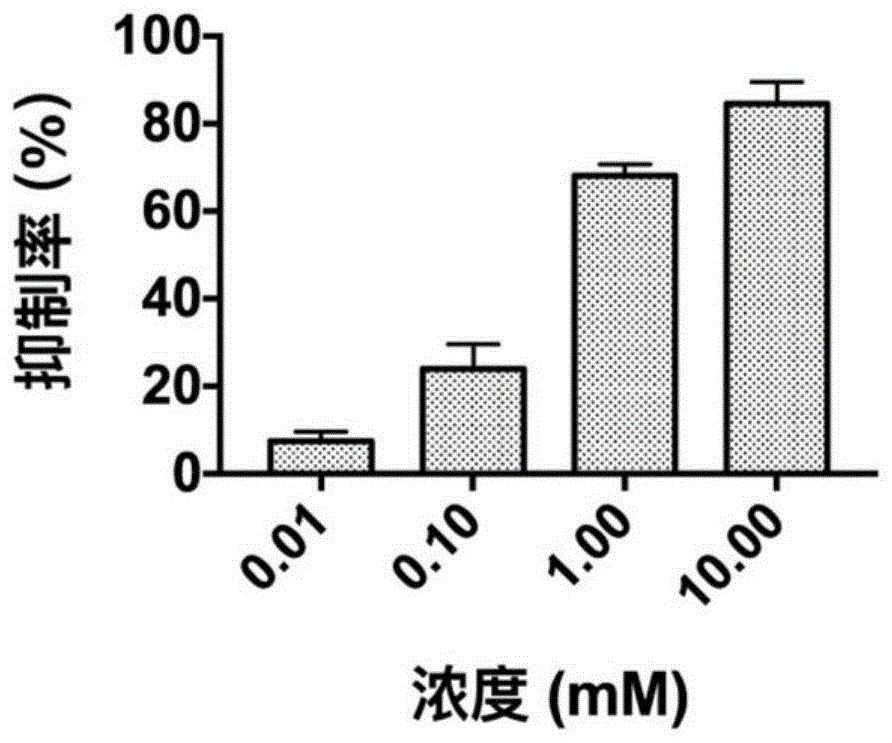
图1为实施例1的小分子抑制剂对人源鸟氨酸脱羧酶的抑制效果。
图2为实施例1采用mtt法检测抑制剂对肿瘤细胞的杀伤效果。
具体实施方式
实施例1
所涉及的苯甲酰胺小分子抑制剂为:2,3,4,5,6-五氟-n-(3-氟烯基)苯甲酰胺,结构式如图所示。
活性检测方法如下:
1.人源odc原核表达质粒的构建
将人源odc的基因序列,通过bamhi和xhoi酶切位点,插入到pet28a质粒中,构建出pet28a-hodc质粒,经dna测序验证。
人源odc的基因序列:
atgaacaactttggtaatgaagagtttgactgccacttcctcgatgaaggttttactgccaaggacattctggaccagaaaattaatgaagtttcttcttctgatgataaggatgccttctatgtggcagacctgggagacattctaaagaaacatctgaggtggttaaaagctctccctcgtgtcacccccttttatgcagtcaaatgtaatgatagcaaagccatcgtgaagacccttgctgctaccgggacaggatttgactgtgctagcaagactgaaatacagttggtgcagagtctgggggtgcctccagagaggattatctatgcaaatccttgtaaacaagtatctcaaattaagtatgctgctaataatggagtccagatgatgacttttgatagtgaagttgagttgatgaaagttgccagagcacatcccaaagcaaagttggttttgcggattgccactgatgattccaaagcagtctgtcgtctcagtgtgaaattcggtgccacgctcagaaccagcaggctccttttggaacgggcgaaagagctaaatatcgatgttgttggtgtcagcttccatgtaggaagcggctgtaccgatcctgagaccttcgtgcaggcaatctctgatgcccgctgtgtttttgacatgggggctgaggttggtttcagcatgtatctgcttgatattggcggtggctttcctggatctgaggatgtgaaacttaaatttgaagagatcaccggcgtaatcaacccagcgttggacaaatactttccgtcagactctggagtgagaatcatagctgagcccggcagatactatgttgcatcagctttcacgcttgcagttaatatcattgccaagaaaattgtattaaaggaacagacgggctctgatgacgaagatgagtcgagtgagcagacctttatgtattatgtgaatgatggcgtctatggatcatttaattgcatactctatgaccacgcacatgtaaagccccttctgcaaaagagacctaaaccagatgagaagtattattcatccagcatatggggaccaacatgtgatggcctcgatcggattgttgagcgctgtgacctgcctgaaatgcatgtgggtgattggatgctctttgaaaacatgggcgcttacactgttgctgctgcctctacgttcaatggcttccagaggccgacgatctactatgtgatgtcagggcctgcgtggcaactcatgcagcaattccagaaccccgacttcccacccgaagtagaggaacaggatgccagcaccctgcctgtgtcttgtgcctgggagagtgggatgaaacgccacagagcagcctgtgcttcggctagtattaatgtgtag
我们使用的上述人源odc序列,与数据库(http://www.ncbi.nlm.nih.gov/nuccore/nm_002539.1)中的序列有一个碱基变化(方框标记的c在数据库序列中为t,对应的氨基酸由精氨酸变为半胱氨酸),但不影响其活性。
2.人源odc蛋白的表达
将上述pet28a-hodc质粒通过cacl2法,转化到大肠杆菌bl21菌株中,通过卡那霉素进行筛选。将在含有卡那霉素的luria-bertani(lb)培养板上生长的菌株,接种至含有卡那霉素的lb液体培养基中,37℃、250rpm培养至对数生长期,然后添加iptg(异丙基硫代半乳糖苷)至0.5mm,在28℃诱导表达4小时。最后,离心收集细菌。
3.人源odc蛋白的纯化
将上述收集的细菌,用裂解液(50mmtris/hcl,ph8.0,300mmnacl,1mmdtt,1mmpmsf,5mmimidazole.)重新悬浮,然后通过超声方法进行细胞裂解。然后将裂解液在4℃、12000转离心后,保留上清。将上清利用ni-ntahis标签蛋白结合填料进行结合和纯化,人源odc蛋白的洗脱缓冲液为50mmtris/hcl,ph8.0,300mmnacl,1mmdtt,100mmimidazole。
4.人源odc蛋白的活性检测
在1.5ml的ep管中,加入400ul底物反应混合物(在150mmpbs(ph7.1)中溶解17.57ulβ-巯基乙醇,55.84mg1.5mmedta二盐钠,75nmplp储液,2mm鸟氨酸盐酸盐)。然后,加入50ug的odc蛋白。混匀后将ep管放置在37℃水浴中30min。然后加入400ul10%的三氯乙酸终止反应。室温离心5000rpm,5min。取出100ul上清,与200ul4mol/l的naoh溶液混合,再加入400ul正戊醇,充分混合。2000rpm离心5min,将上层液200ul转移至新的ep管中,加入200ul四硼酸钠(0.1mol/l,ph8.0)充分混合。然后加入200ul三硝基苯磺酸(10mmol/l),充分混合。再加入400uldmso,充分混合1min。3000rpm离心5min。取出上层液到96孔板中,用酶标仪检测426nm处的吸光值。
5.抑制剂对人源odc蛋白的抑制活性的检测
在上述活性检测步骤中,加入400ul底物反应混合物后,随即加入小分子药物并混合。后续步骤一样。
通过以下公式计算出odc抑制率
对照差=加酶对照组平均od值-未加酶对照组平均od值
实验差=加酶实验组平均od值-未加酶实验组组平均od值
抑制率=(对照差-实验差)/对照差×100%
按照上述方法得到的该小分子抑制剂的抑制效果如图1所示,从图中可以看出在50%以上抑制的浓度时,该药物能抑制odc的活性,其抑制能力与2.5mm的dfmo相当。
6.采用mtt法检测抑制剂对肿瘤细胞的杀伤效果
采用mtt法检测抑制剂对肿瘤细胞的杀伤效果
将a549细胞接种至96孔细胞培养板中,接种密度2000个细胞/孔,培养基为rpmi-1640,10%小牛血清,1%链/青霉素,体积100ul/孔。在37℃细胞培养箱中培养24小时后,加入100ul用rpmi-1640稀释的小分子药物或培养基。继续培养60小时后,吸去培养基,加入100ulmtt(终浓度250ug/ml)后,继续培养4小时。然后吸去培养液,加入150uldmso,在摇床上低速振荡15min,使结晶物充分溶解。最后,利用酶标仪读取490nm处的吸光值。
按照上述方法得到的mtt法检测抑制剂对肿瘤细胞的杀伤效果如图2所示,从图中可以看出在在50%以上抑制的浓度时,该药物能够抑制和杀伤肿瘤细胞,但在50%以下抑制的浓度时能力很弱。
- 还没有人留言评论。精彩留言会获得点赞!
- SPNP‑I在制备Nav1.7通道工具试剂中的应用的制造方法与工艺
- SPNP‑I在制备Nav1.2通道工具试剂中的应用的制造方法与工艺
- SPNP‑IX在制备Nav1.3通道抑制剂中的应用的制造方法与工艺
- SPNP‑IX在制备Kv4.2通道工具试剂中的应用的制造方法与工艺
- SPNP‑IX在制备Kv4.3通道工具试剂中的应用的制造方法与工艺
- SPNP‑27在制备Nav1.5通道抑制剂中的应用的制造方法与工艺
- SPNP‑27在制备Nav1.3通道抑制剂中的应用的制造方法与工艺
- 一种多肽在制备Nav1.3钠通道工具试剂中的应用的制造方法与工艺
- SPKP‑XI在制备Kv4.2通道抑制剂中的应用的制造方法与工艺
- SGLT‑2糖尿病抑制剂及其中间体的制备方法与流程