基因组/蛋白质组序列的表示、可视化,比较以及报告的制作方法
以下涉及生物信息学、基因组处理技术、蛋白质组处理技术、以及相关技术。
背景技术:
基因组或蛋白质组数据包括碱基或肽的有序序列。在脱氧核糖核酸(dna)的情况下,碱基是腺嘌呤、胞嘧啶、鸟嘌呤和胸腺嘧啶,它们通常分别由字母“a”、“c”、“g”、和“t”表示。在核糖核酸(rna)的情况下,碱基是腺嘌呤、胞嘧啶、鸟嘌呤和尿嘧啶,它们通常分别由字母“a”、“c”、“g”和“u”表示。dna和rna不同在于由尿嘧啶(u)取代胸腺嘧啶(t)。
蛋白质和其他蛋白质组分子包括通过肽键连接的氨基酸。相应的蛋白质组数据适宜由肽(或氨基酸)序列表示。(术语“肽”和“氨基酸”在本文中可互换使用来指蛋白质组序列的元件)。氨基酸通常是由一个字母或三个字母代码来表示。例如:丙氨酸是由字母“a”或三个字母代码“ala”表示;精氨酸是由字母“r”或三个字母代码“arg”表示;等等。在肽序列中,由于肽键不会改变,各个肽通常由它们的氨基酸成分表示。因此,例如,“a”或“ala”用于在蛋白质序列中表示包含丙氨酸的肽。
基因组或蛋白质组数据中包含大量的有用信息,其通常通过模式匹配来提取。例如,基因组或蛋白质组数据可以查找到在过去的临床研究中已经与某种疾病相关联的疾病标记,或者该数据可以被用于某些疾病的治疗计划,如癌症、代谢障碍、等等。指示祖先谱系的遗传标记可被用于评估受试者的祖先。在执法或某些其他领域,表型与基因型的关联可以提供有用的信息。例如,从犯罪现场获得的dna样本可能表明罪犯有某物理特征,从而排除不具有该特征的任何疑犯。
序列处理通常包括以下操作:获取序列片段;序列片段相对于参考序列的比对(例如,一些合适的参考序列包括:refseq、hgl8、hgl9、泛基因组、等等);以及对比对后的序列进行分析来识别特定主题的变化。在没有参考映射(map)时其可能还包括从头比对,导致多种长度的重叠群的产生,然后可以将0其注释和进行比较分析。
基因组或蛋白质组数据通常被获取为序列片段,其以标准化的格式存储,例如fasta或fastq。一种合适的输出fasta或fastq数据的序列分析仪系统线是illumina测序仪(购自illumina公司,圣地亚哥,加利福尼亚州,美国)。fasta格式使用单个的字母来代表每个碱基或肽(例如,用于dna的碱基表示“a”、“c”、“g”和“t”或用于rna的“a”、“c”、“g”和“u”)。额外的单个字母代码可被提供以表示不明确的碱基—例如,字母“r”可以表示不明确的碱基,腺嘌呤或鸟嘌呤。fastq格式是fasta的扩展,它包括表示该碱基序列的质量值的字符的附加线。由于为每个碱基使用了两个字符(一个字符来表示碱基值,第二个字符表示质量值),fastq文件大约两倍于相应的fasta文件。
该序列片段相对于一个参考序列被比对以产生比对后的基因组或蛋白质组数据,其通常被存储在序列比对/染色体图(sam)文本文件或等效的二进制bam文件中。sam格式通常采用“基于1”的坐标系统,其中序列的第一个碱基的坐标是一,而bam格式通常使用一种“基于0”的坐标系统,其中序列的第一个碱基的坐标是零。该序列片段形成的比对后的序列适宜保持在fasta格式(在bam的情况下转换为二进制格式)。序列比对允许识别功能区域,例如,基因、内含子(基因内的不翻译成蛋白质的不相干的子序列)、外显子(基因的被翻译成蛋白质的部分)、启动子(促进基因转录的子序列)、转录因子(tf)结合位点的编码序列(与dna序列中tf结合位点相结合的tf蛋白,以控制转录)、转录到非编码rna(nc-rna)的序列、等等。
比对后的基因组或蛋白质组序列然后被分析以识别变化,如单核苷酸多态性(snp)、拷贝数变异(cnv)、子序列的插入或缺失(indel)特性、各种染色体内和/或染色体间重排等等。这些变化可作为疾病的标志、祖先谱系标记等等。
序列处理是计算密集型的,并产生大量的产品数据。为了便于说明,一个典型的基因组测序研究可能产生大约40gb的fasta数据,和/或大约80gb的fastq数据。这些序列片段的比对产生的大约200gb的一个或多个sam文件(以二进制bam格式可减少至约100gb)。比对后的数据由模式匹配算法处理来识别感兴趣的变化,并且这些研究的结果通常存储为各种文本文件、表格、电子表格或其它数据汇编。
这些现有的方法具有某些缺点。医生或其他审阅者可能需要访问和检查大量的文件以获得所需的信息。变异分析本身也可以由于数据存储的多样性而变得复杂。例如,考虑一个发生在外显子的变化,而不是其他地方,是有证明力的。在变分分析中,首先进行模式匹配,以识别与变化签名相匹配的候选序列。此后,访问源sam文件以确定该候选序列匹配是否发生在外显子中。这个变异分析包括两个步骤:(1)匹配碱基序列;以及(2)匹配碱基序列匹配到外显子。(可替代地,外显子数据可以使用sam内容首先被识别,随后碱基序列匹配仅被应用到外显子数据。然而,又一次地,这是一个两步的过程)。
测序结果的呈现也由于数据存储的多样性而变得复杂。通常情况下,每个变异分析的结果都存储在自己的文本文件、表格、电子表格或其他数据汇编中。因此,医生或其他审阅者需要检查不同的数据汇编来访问研究的结果。这个过程可能会错过信息组合的协同或不和谐。表和/或电子表格的文本格式也可能很难理解。一种解决方案是附加地以图、彩色编码图、或类似的形式呈现选定的结果。然而,这种辅助的数据表示的生成进一步提高了计算复杂度。此外,图形化的结果摘要可将这些结果和底层的基因组或蛋白质组序列数据分离。
技术实现要素:
下面提供了此处公开的新的和改进的装置和方法。
根据一个公开的方面,一种方法包括:编码基因组或蛋白质组数据为基因组或蛋白质组字符串,其包括生物信息学字符集中的字符,其中:(i)基因组或蛋白质组数据的每个碱基或肽由生物信息学字符集的一个单个的字符表示,以及(ii)生物信息学字符集的每个字符编码(i)碱基或肽和(ii)与碱基或肽相关的至少一个带注释的数据值;以及通过使用映射到生物信息学字符集的生物信息学字体显示基因组或蛋白质组字符串来显示基因组或蛋白质组数据。该编码和显示被适当地由数字处理设备执行。该方法可以进一步包括对基因组或蛋白质组字符串执行至少一个串函数来生成更新的基因组或蛋白质组字符串,在其中至少一个碱基或肽由编码了至少一个由执行的字符串操作生成的附加的或修改的注释的基准的一个单个的字符表示。
根据另一个公开的方面,一种数字处理设备被配置为执行在上一段中陈述的方法。根据另一个公开的方面,一种非临时性(non-transitory)存储介质是由数字处理设备可读的,并存储由数字处理设备可执行的指令来执行在上文中所述的方法。
根据另一个公开的方面,一种非临时性存储介质是可由数字处理器读取的,并存储用于处理表示为包括生物信息学字符集的字符的基因组或蛋白质组字符串的基因组或蛋白质组数据的软件,其中基因组或蛋白质组数据的每一个碱基或肽由生物信息学字符集的单个字符表示,并且生物信息学字符集的字符编码碱基或肽以及与碱基或肽相关的附加数据。在一些实施例中,所述软件使用字符串处理运算(operation)来处理基因组或蛋白质组数据。在一些实施例中,所述软件使用逐位掩码运算(bitwisemaskingoperation)处理所述基因组或蛋白质组数据以将表示碱基或肽的字符的选择的二进制位设置为零。在一些实施例中,所述存储介质进一步存储映射到生物信息学字符集的生物信息学字体,以及上述软件执行显示运算,在其中使用生物信息学字体显示基因组或蛋白质组数据。
一个优点在于基因组或蛋白质组数据更加紧凑的和集中存储。
另一个优点在于存储了碱基质量值、功能区域信息,变化信息、或者对以紧凑的单字符表示的碱基或肽序列的其他注释数据。
另一个优点在于提供了碱基或肽序列的直观的显示,包括表示或描绘注释信息,例如碱基质量值、功能区域、变化等等,的变音符或其他字体特征。
另一个优点在于使用常规的字符串运算比较基因组序列。字符串的比较可容易地配置为只检测注释中的特定的变化(例如,甲基化)。
其它优点包括便于对由不同的研究小组注释的参考序列的不同注释的比较,并使正常对比癌症基因组的后生变化的可视化变得容易。
在阅读和理解下面的详细描述后,进一步的优点对本领域的普通技术人员来说是显而易见的。
附图说明
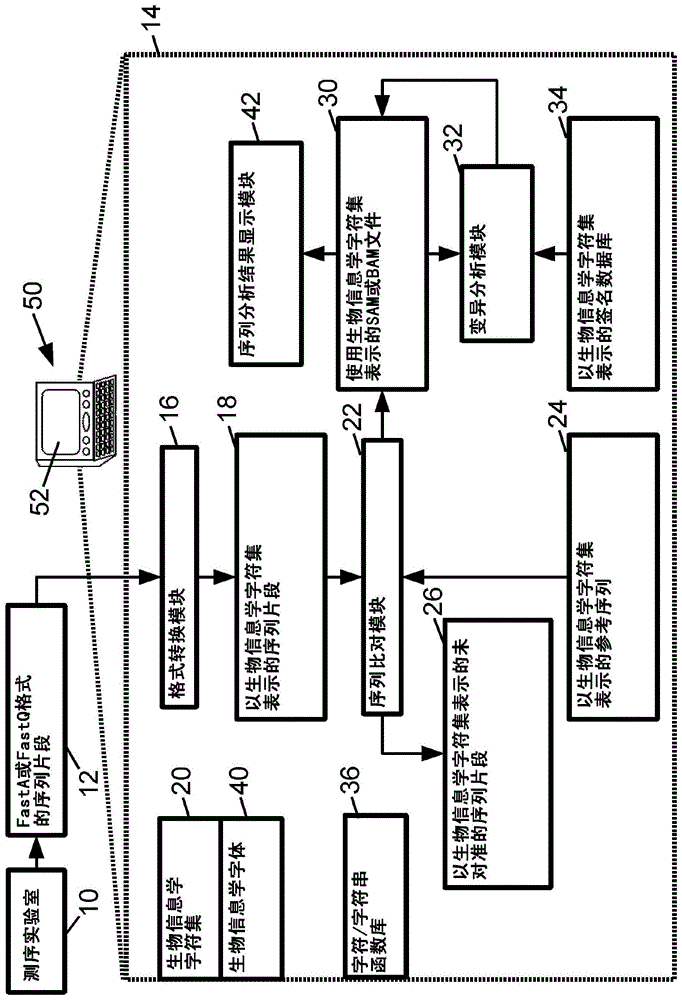
图1示意性地示出了采用生物信息学字符集和对应的生物信息学字体的序列比对/分析模块。
图2以表格形式列出了随着碱基序列描绘碱基的质量分数的合适的区别标记(diacriticalmarking)系统。
图3示出了碱基序列的一部分的描绘,包括描绘编码序列(cds)的开始和结束密码子的区别标记。
图4以表格形式列出了描述注释碱基的区域和变化信息的一些合适的区别标记。
图5示出了碱基序列的一部分的描绘,包括描绘位于两个外显子之间的内含子的选定的区别标记。
图6示意性地显示了通过使用按位或运算结合用于碱基的位图和用于区别标记的位图来构建生物信息学字体的字体字符的方法。
图7示出了碱基序列的一部分的描绘,包括使用图6的实施例的生物信息学字体的区别标记描绘编码序列(cds)的开始和结束密码子的区别标记以及描绘内含子的区别标记。
图8示意性地示出了碱基序列的一部分,其与复选框一起显示,通过它审阅者可以选择哪些注释类型要被描绘在显示的碱基序列中。
图9示意性地显示了一种通过使用按位或运算结合用于氨基酸的位图和用于区别标记的位图构建用于表示氨基酸以及它们的生物物理特性的生物信息学字体的字体字符的方法。
具体实施方式
参照图1,基因组或蛋白质组测序分析系统包括测序实验室10,其准备并测序dna、rna、蛋白质等等的样本,以产生以fasta或fastq格式12被存储为序列片段的碱基序列片段(对于基因组样本)或肽序列片段(对于蛋白质组样本)。通过说明性示例的方式,dna材料的样本可通过诸如dna的纯化和克隆的操作来准备和测序,通过诸如使用荧光标记的链终止子如双脱氧核苷酸三磷酸(ddntp)终止子的聚合酶链反应(pcr)的技术被放大,并通过细管电泳或其他测序技术测序。在实验室10这些不同的操作可以通过手动、半自动或全自动的处理操作来执行。例如,某些样本准备操作可以手动执行或以半自动的方式进行,接着所准备的样品的装载和测序使用自动测序仪。附加地或可选地,实验室10可以使用埃德曼(edman)降解和质谱分析法或其它合适的技术执行蛋白质组测序。
说明性的测序实验室10的输出包括以fasta或fastq格式12的序列片段。这些是传统的格式。在fasta中,碱基或肽的序列由字符序列表示,其中每个字符代表一个碱基或肽。例如,基因组序列“腺嘌呤—鸟嘌呤—胞嘧啶”以fasta被适当地表示为“agc”。在fastq格式中,平行于代表碱基或肽的字符串的第二字符串被添加。该第二字符串表示了使用单个字符代码的每个碱基或肽的质量值。因此,以fastq每个碱基或肽由2个字符表示:第一(例如,碱基序列)字符串中的第一字符给出了碱基或肽的标识,第二(例如,质量值)字符串中的第二字符给出了由自动测序仪(或其它合适的质量评估)输出的该碱基或肽的质量值。
继续参考图1,序列比对/分析模块14接收序列片段12。格式转换模块16将fasta或fastq序列转换为采用生物信息学字符集20的单个字符串表示18。如本文所公开的,生物信息学字符集20被设计来以紧凑的单个字符串格式表示基因组或蛋白质组序列,其中每个基碱基或肽通过生物信息学字符集20的单个字符来表示。该单字符编码碱基或肽以及与碱基或肽相关的注释数据。
在此处认识到现有的格式如fasta是不紧凑的。例如,考虑到编码为基因组数据。为了编码四个碱基(假设胸腺嘧啶或尿嘧啶,但不能同时)需要四个编码值。这四种可能性可以使用尽可能少的两位被编码。如果不确定也被编码,那么这可能需要多达15个可能的值:四个“已知”的碱基,另外六个不确定的“双碱基”的组合(例如,已知是腺嘌呤或胞嘧啶的一个位置),四个可能的不确定的“三碱基”的组合(例如,已知是腺嘌呤,胞嘧啶,或鸟嘌呤的一个位置),和一个完全不确定的组合(也就是,可以是腺嘌呤,胞嘧啶,鸟嘌呤,或胸腺嘧啶的一个位置)。这15个可能的值可以只使用4位被编码。如果不太详细的不确定是要被编码的,需要更少的可能值。例如,如果只有4个碱基和单个不确定的“n”代码被使用时,则只有5个可能值,其可使用3位进行编码。然而,fasta采用一个完整的字节(8位(bit))来表示信息。
fasta的单字节编码方案确实有巨大的优势。单字节的方法适应于典型的数字处理器架构,在其中数据被组织成每八位一个的字节单位。此外,“a”、“c”、“g”、“t”字符(以及可选的不确定字符,如“n”),符合美国信息交换标准码(ascii)字符集,因此现有的字符串函数可以被用来操纵fasta数据。但是,在此处认识到,fasta“浪费”了大量的每个字节的编码能力。一个字节的8位可存储256个可能的值(范围从0到255),而fasta仅使用15个(或更少)可能的组合。
fastq是fasta的扩展。一个fastq格式文件的碱基编码(或肽编码)字符串与fasta是相同的,因此上述评论也适用于fastq。此外,fastq包括包含碱基质量值的第二字符串,一个字节被用于每个质量值。如果phred质量分数被编码,这些分数范围从0到93。这94个可能的值可以用尽可能少的7位编码,但fastq使用一个完整的8位字节来编码质量值。
相反,此处公开的生物信息学字符集20的实施例提供更紧凑的存储,其具有更多的优点。该生物信息学字符集20使用单个字符来表示序列中的每个碱基或肽。为了保留fasta和fastq与现有的数字处理架构相适应的优势,每个字符通常是一个单字节或两字节。设计该生物信息学字符集20为每个字符使用单个字节意味着该字符集很好地与同样为每个字符采用单个字节的标准ascii相适应。另一方面,生物信息学字符集20为每个字符使用2个字节的实施例很好地与为每个字符使用两个字节的标准unicode相适应。
然而,该生物信息学字符集20不是ascii或unicode。相反,该生物信息学字符集20被设计成使用字符的位的一个子集来表示碱基或肽,并使用字符的位的另一个子集来表示与碱基或肽相关联的至少一个注释的数据值(通常几个不同的注释的数据值)。
通过说明性示例的方式,表1中列出了该生物信息学字符集20的实施例,其适合于表示基因组数据(更具体的,dna序列),每个碱基使用单个字符,每个字符是具有16位的2字节字符。单个字符的位被适当地写成b15b14b13b12b11b10b9b8b7b6b5b4b3b2b1b0,其中b15是最高有效位,b0是最低有效位。最高有效字节包含位b15b14b13b12b11b10b9b8而最低有效字节包含位b7b6b5b4b3b2b1b0。在表1的生物信息学字符集表20中,位b2b1b0被用来代表碱基。有五个可允许的值:4个值来表示dna的4个碱基,以及第五个“不确定”的值表示一个未知的碱基。剩余的13位b15b14b13b12b11b10b9b8b7b6b5b4b3被用来代表与碱基相关联的各种注释的数据值。(见表1的说明性的例子)。
在表1的生物信息学字符集20的一个示例性的实施例的情况下,由格式转换模块16执行的格式转换适当地操作如下:fasta代码“a”转换为字符0000000000000000bin(0000hex);fasta码“c”转换为字符000000000000000lbin(000lhex);fasta码“g”转换为字符0000000000000010bin(0002hex);fasta码“t”转换为字符000000000000001lbin(0003hex),以及所有其他与不确定的碱基对应的fasta码转换为字符0000000000000100bin(0004hex)。在本实施例中fastq以同样的方式被转换,除了位b6b5b4b3也被填充了对应于表1中所示的编码方案的质量值的编码。请注意,这种编码方案使phred分数只由四位表示,尽管有一些准确度的损耗(例如,b6b5b4b3=0100指定phred分数的取值范围为31—40)。通常情况下,人们只是想知道,质量分数是否为“高”或“低”,所以这个质量值准确度的损耗一般是不会有问题的。
在表1的生物信息学字符集20的一个示例性的实施例的情况下,由格式转换模块16执行的转换设置剩余的注释位b15b14b13b12b11b10b9b8b7为默认值零。这反映了比对和变异分析尚未进行,因此不存在识别为属于外显子、内含子、等等的碱基的现实。
在图1的说明性的实施例中,测序实验室10产生fasta或fastq格式的数据12,然后其通过格式转换模块16被转换为使用生物信息学字符集20的数据表示18。这种方法有利地使得序列比对/分析模块14处理以常规fasta或fastq格式生成的测序片段数据。然而,可替换地,测序实验室10可以直接输出生物信息学字符集20表示的序列的片段。
继续参照图1,以生物信息学字符集20表示的该序列片段18由序列比对模块22处理。所述序列比对模块操作以比对序列片段,通常参考一参考序列24,以将该序列片段“拼接在一起”以形成(多个)完整的和长的(更长的)比对序列。比对处理类似于常规应用于fasta序列片段的方法,即,匹配序列片段的末端以对准并拼接在一起。然而,数据表示18的注释位(例如,表1中的例子中的质量值注释b6b5b4b3),可能会导致错配。换言之,具有不同质量值的两个相同的碱基可能由于不同的质量值而不会匹配。
表1
为了解决这个问题,为了比对处理的目的,注释位的值被适当地设置为零。这可以使用按位“与”掩码来高效地完成,该掩码中注释位的位置被设置为零,碱基位的位置被设置为一。对于表1中的例子中,一个合适的位掩码是mbase=000000000000011lbin(0007hex)。将该掩码应用到生物信息学字符集20的字符c(适合被写为c&mbase,其中符号&表示逐位“与”运算)具有将所有注释位归零,而字符c的碱基位通过不变的效果。二进制掩码是一个低级别的数字处理运算,因此通常是相当有效的。对于由一个生物信息学字符集20的k个字符的字符串s=[c1c2c3...ck]表示的碱基序列,每个字符需要被单独屏蔽,例如使用k次迭代的循环来迭代地轮流应用掩模mbase到每个字符c1,...,ck。为了标记方便,在此处这个字符串掩码运算由s&mbase形式的伪代码表示,其中s是碱基序列字符串,mbase是用于字符串s的单个字符的二进制掩码。因此,为了片段比对的目的而比较碱基序列片段s1和s2(也就是,比较各自的碱基而不考虑注释位),比较在s1&mbaseands2&mbase之间进行。
当序列比对模块22参照参考序列24比对序列片段时,它也可以识别有意义的基因组区域,例如外显子、内含子、启动子区域、编码序列(cds)区域、等等。如果参考序列24使得这些区域被标记或以其他方式被指示,这是可以实现的。在序列比对模块22识别这样的功能区之处,它适当地设置相应的注释位来表示这些区域。因此,例如(再考虑表1中的示例性的生物信息学字符集),如果给定碱基被识别为内含子的一部分,则位b10被设置为1。如果一个给定的碱基被识别为外显子,则位b9被设置为1。
注意,该序列比对模块22可以仅更新编码由该比对过程识别的基因组区域的注释位。另一方面,序列比对模块22不能更新编码不是由比对确定的变化信息的注释位。例如,该序列比对模块22不能更新编码碱基是否是疾病标志的一部分的位b15。
在一些情况下,序列比对模块22可能无法拼接一些序列片段为对准的序列。这些剩余的未对准的序列片段可以是来自在克隆过程中使用的宿主细胞的剩余的dna产物,或可以反映测序处理中的错误,或者可能归因于其他因素。这些剩余的未对准的序列片段被适当地存储为一个数据结构26,未对准的片段也使用生物信息学字符集20来表示。
对准的序列,它是序列比对处理的期望产物,被适当地存储在序列比对/染色体图(sam)文件或等效的二进制bam文件30中。然而,在sam文件中,对准的序列使用生物信息学字符集20合适地表示。有利的是,这意味着注释信息,如碱基质量值(在表1的例子中,注释位b6b5b4b3)和功能区域信息(在表1的例子中,注释位b14b13b12b11b10b9b8b7)被和基因组序列自身中的碱基一起直接存储在sam或bam文件30中。
对准的序列由变异分析模块32处理,其执行一个或多个变异分析。该分析通常使用模式匹配运算来执行,其中,对准的序列与从签名数据库34得到的标记模式相比较。该标记模式最好也使用生物信息学字符集20存储。按位掩码可以被用来基于与变异分析的相关性选择性地排除或保留注释位。
例如,考虑一个变异的例子,当它发生在外显子中而不是在其他地方是有证明力的。在这种情况下,碱基是否是外显子的一部分是相关的,但其他的注释(例如,碱基质量分数)是不相关的。在表1的生物信息学字符集20的实施例中,该碱基是否是外显子的一部分由注释位b9指示。因此比较适当地对序列字符串s&m执行,其中位掩码m=0000001000000111bin(0207hex)。该掩码保留了外显子注释位b9和碱基位b2b1b0,将字符的其余位归零。数据库34中的变异签名的碱基具有形式0000001000000xxxbin(020xhex),其中x可以是0或1,x表示000,001,010,011或100的任意一个(在表1的实施例的形式中其余的可能性不编码的任何东西)。因此,s&m和签名之间的比较提供了期望的模式匹配。
有利的是,这种方法使用逐位运算和字符/字符串函数实现了变异分析,并确实可以利用现有的字符/字符串函数库36,如c++、perl或其他编程语言、或脚本语言等等提供的标准字符/字符串库。标准的字符或字符串函数通常被设计为在标准的字符集上操作,如ascii或unicode;然而,如果生物信息学字符集20“适应”于标准字符集(如ascii或unicode),则该字符/字符串函数还将操作生物信息学字符集20。在这样的情况下,如果生物信息学字符集20采用相同的字符大小(例如,一个字节的ascii或两个字节的unicode)并避免任何在特定的数字处理平台上在ascii或unicode中可以具有特别显著的意义的“特殊”字符被使用,生物信息学字符集20“适应”于ascii或unicode。例如,在某些平台上空字符可以被用作ascii字符串的终止,在此情况下,没有生物信息学字符集20的实施例的字符采用与ascii空字符具有相同的数字代码的单字节字符。此外,在这个上下文中,生物信息学字符集20“适应”于ascii或unicode意味着以生物信息字符集20书写的字符串以和ascii或unicode字符串在特定数字处理平台上被使用的同样的方式被格式化,以便良好的形成输入到标准的字符串函数。例如,在某些平台上,unicode字符串可以包括两个字节的头部,其指示字符是否是大端(通常头部fehex,ffhex)或小端(通常头部ffhex,fehex)。在这种情况下,在被输入到一个为unicode设计的标准字符串函数之前,适当的2字节的头部应该作为前缀加到以生物信息学字符集20表示的字符串中。
当变异分析模块32识别所关注的变异,它适当地设置相应的注释位来表示该变异。因此,例如(再考虑表1中的示例性的生物信息学字符集),如果在基因组序列中一个疾病标志被识别,然后将每个匹配该疾病标志的碱基的位b15被设置为1,该更新在sam(或bam)文件30中完成。由于序列比对模块22已经更新基于比对处理识别的注释位,并包括这些注释在sam(或bam)文件30中,跟随的是,一旦变异分析完成,sam(或bam)文件30将包括以单一的紧凑表示的所有相关的功能区域标识,与变异相关的信息,以及碱基质量值。
当序列处理(例如,比对和变异分析)完成时,其结果可以人类可感知的形式被显示(例如,显示在显示设备上,通过打印机或其他标记引擎打印,等等)。通常,这种显示包括使用传统的符号(例如,用于dna的碱基序列的字母“a”、“c”、“g”和“t”的组合)列出该序列,并提供报告总结变异分析结果。代替使用字母,在某些系统中其它符号被用于碱基。例如,在一个约定中,腺嘌呤被表示为符号“×”,胞嘧啶被表示为符号“□”,鸟嘌呤被表示为符号“+”和胸腺嘧啶被表示为符号“”。生成序列列表通常是直接的,因为fasta或fastq序列串采用ascii码的一个子集。也就是说,在ascii和fasta(或fastq)两者中字母“a”是由41hex表示的;在ascii和fasta(或fastq)两者中字母“c”是由43hex表示;在ascii和fasta(或fastq)两者中字母“g”是由47hex表示的;在ascii和fasta(或fastq)两者中字母“t”是由54hex表示的。因此,fasta(或fastq)碱基字符串是ascii字符串,并且可以使用任何字体映射为ascii被打印。
在另一方面,序列比对/分析模块14使用与ascii(或unicode)不同的生物信息学字符集20,虽然它最好与ascii(或unicode,对于2字节字符集的实施例)相适应。因此,虽然使用生物信息学字符集20表示的基因组或蛋白质组数据可以使用映射到ascii(或双字节字符的实施例的unicode)的标准字体被正式地打印,但所显示的数据似乎是无意义的。因此,该序列比对/分析模块14包括映射到的生物信息学字符集20的生物信息学字体40。该字体40提供为生物信息学字符集20的每个字符被显示的字体字符。序列分析结果显示模块42适当地使用生物信息学的字体40显示以生物信息学字符集20表示的基因组或蛋白质组序列。生物信息学字体40的字体字符最好包括:(1)一个字母来表示碱基或肽(或可选地三个字母的序列来表示肽)和(2)附加的特征,如区别标记,字体样式方面,例如粗体和/或斜体字体样式等等,来表示与在生物信息学字符集20的字符表示的碱基或肽相关的注释数据。作为使用字母的替换,碱基或肽的表示(1)可以采用另一种类型的符号,如:腺嘌呤=“×”,胞嘧啶=“□”,鸟嘌呤=“+”和胸腺嘧啶=“”。优选地,所述序列分析结果显示模块42可以利用由平台提供的常规的显示ascii或unicode文本的文本显示例程。这些文本显示例程适应于通过调用显示程序使用生物信息学字体40显示(或打印)来简单地显示基因序列。
序列比对/分析模块14可以被实现为一个数字处理设备,例如一个说明性的计算机50,它包括一个数字处理器(未示出),其被编程来执行实现各个模块16、22、32的软件,并包括存储器存储生物信息学字体40。除了说明性的计算机50,另一种数字处理设备也可被使用,例如专用的dna测序装置,它包括一个数字处理器,或一个网络服务器系统,或一个图形处理单元(gpu),诸如游戏机,其被重新编程以实现序列比对/分析模块14等等。序列比对/分析模块14可选地包括或访问一个显示设备(诸如计算机50的说明性的显示器52),其用于显示信息,诸如使用生物信息学字体40表示的基因组或蛋白质组序列。
序列比对/分析模块14还可以被实现为一个非临时性存储介质,其存储生物信息学字体40和软件,当该软件由一个数字处理器(例如,计算机50的处理器)执行时实现各个模块16、22、32。这样的非临时性存储介质可以通过说明性示例的方式,包括下列一项或多项:一个硬盘驱动器或其它磁性存储介质;一个光盘或其他光存储介质;一个只读存储器(rom),随机存取存储器(ram),闪存,或其他静电存储器或静电存储器的组合;等等。
图解的图1示出的序列比对/分析模块14概略地表示生物信息学字符集20。然而,可以理解的是,在一些实施例中生物信息学字符集20通过各个模块16、22、32执行的操作被隐含实现,这些模块创建和操作使用生物信息学字符集20格式化的基因组或蛋白质组序列。另一方面,生物信息学字体40被存储为位图的集合或其他字体字符表示。在一些实施例中可以预期通过结合或修改存储的构成特征位图来为给定的字体字符“按需”构建位图,例如,通过添加表示注释数据的一个或多个区分标记到描绘表示碱基或肽的字母的位图。
已经参照图1和在表1中给出的生物信息学字符集20的实施例描述了示例性的序列比对/分析模块14,生物信息学字符集20的一些另外的实施例和一些说明性的生物信息学字体字符将在下面被描述。
参照图2和表1,一些适用于显示生物信息学字符集20的字符字体被显示,其包括碱基和碱基质量值的信息两者。在这种方法中,表示碱基质量值的变音符号从碱基质量注释位b6b5b4b3确定,并包括一组一个或多个连接的(如图2所示)或分离的线段,其总长度指示碱基的质量值。请注意,在图2的实施例中,变音注释值1000bin,1001bin和1010bin都被映射到相同的字体字符。因此,该字体字符表示一个大于70的phred得分。图2中的区别标记显示由字母“a”指示的碱基腺嘌呤。更一般地,在一个合适的实施例中,碱基腺嘌呤是由字母“a”或“a”指示的;碱基胞嘧啶是由字母“c”或“c”指示的;碱基鸟嘌呤是由字母“g”或“g”指示的;碱基胸腺嘧啶是由字母“t”或“t”指示的;碱基尿嘧啶是由字母“u”或“u”指示的。图2中的区别标记(即,线)很容易应用到任意的字母。有利的是,审阅者阅读由图2所示的字体字符表示的碱基能够很容易地评估序列中每个碱基的质量值。
应当指出的是,如本文使用的,术语“字母'a'或'a'”表示可以被普通人所理解的、被识别为字母“a”或“a”的符号。字母“a”或“a”可以被不同地表示,例如使用arial类型的外观,或者timesnewroman类型外观,或courier类型的外观,或者手写类型的外观,等等。类似的解释适用于以生物信息学字体40的表示碱基或肽的其他字母。
参照图3和表1,字体被示出用于表示编码序列(cds)启动子和终止密码子。在表1的字符集实施例中,分别通过注释位b7和b8碱基被注释为cds启动子或cds终止密码子。在图3的字体实施例中,b7或b8中任意一个等于1的字符被映射到包括围绕表示碱基的字母的盒子的字体字符;然而,b7和b8两者等于0的字符被映射到不包括这样的盒子的字体字符。如在图3中可见的,这个字体映射的结果是,启动子和终止密码子通过变音盒标记而很容易识别。在图3的实施例中,相同的变音盒标志被用于cds启动子和cds终止子两者,可选的,不同的区别标记可被用于cds启动子和cds终止子,这可能有助于审阅者分辨cds的开始与结束。
参考图4,一些其他合适的用于指示不同基因组区域或变异的区别标记被显示。在图4的例子中,生物信息学字符集20的、包括指示甲基化的注释的字符被映射到生物信息学字体40的包括区别标记的字符字体,该区别标记包括角度标记(即,像“v”,但可选的“v”的点不是朝向下)。在图4所示的另一个例子中,生物信息学字符集20的、包括指示内含子的注释(例如,在表1的例子中b10被设置为1)的字符被映射到生物信息学字体40的包括区别标记的字符字体,该区别标记包括字母“i”或“i”。在图4所示的另一个例子中,生物信息学字符集20的、包括指示外显子的注释(例如,在表1的例子中b9被设置为1)的字符被映射到生物信息学字体40的包括区别标记的字符字体,该区别标记包括字母“e”或“e”。在图4所示的另一个例子中,生物信息学字符集20的、包括指示启动子的注释(例如,在表1的例子中b11被设置为1)的字符被映射到生物信息学字体40的包括区别标记的字符字体,该区别标记包括字母“p”或“p”。在图4所示的另一个例子中,生物信息学字符集20的、包括指示转录因子(tf)结合位点的注释(例如,在表1的例子中b12被设置为1)的字符被映射到生物信息学字体40的包括区别标记的字符字体,该区别标记包括字母“x”或“x”。在图4所示的另一个例子中,生物信息学字符集20的、包括指示非编码rna区域的注释(例如,在表1的例子中b13被设置为1)的字符被映射到生物信息学字体40的包括区别标记的字符字体,该区别标记包括“~”。(在其它设想的实施例中,包括字母“nc”或“nc”或“nc”的区别标记被考虑用于指示nc-rna)。在图4所示的另一个例子中,生物信息学字符集20的、包括指示微rna区域的注释(例如,在表1的例子中b14被设置为1)的字符被映射到生物信息学字体40的包括区别标记的字符字体,该区别标记包括“~”。(在其它设想的实施例中,包括字母“mi”或“mi”或“mi”的区别标记被考虑用于指示mi-rna)。在图4所示的另一个例子中,生物信息学字符集20的、包括指示碱基是疾病标志的一部分的注释(例如,在表1的例子中b15被设置为1)的字符被映射到生物信息学字体40的包括区别标记的字符字体,该区别标记包括“#”或一些其他指定来指示疾病标志的区别标记。附加地或可选地,疾病标记的注释可以通过使用指定的疾病标记的字体样式,如斜体字体样式、粗体字体样式,或粗体斜体字体样式描绘碱基字母(例如,腺嘌呤情况下的“a”)来表示。
图5示出了在描绘的序列区域中这样的字体字符的功效的一个例子。图5示出使用图4的字体表示的dna序列的一部分。由两个外显子区域62、64为界的内含子域区60基于分别指示碱基属于内含子和外显子的区别标记“i”和“e”是很容易辨别。
图4和图5的区别标记仅仅是说明性的例子,其他区别标记以及其他字体特征,如字体样式,字体大小等等也可以被用在生物信息学字体40中来表示不同的注释。例如,在另一种设想的方法中,碱基质量值由描绘碱基的字母的大小来描绘,具有较大的字母表示较高的碱基质量值。这种方法的期望是,审稿人可能会自然地将较小的字母与较低的碱基质量相关联,即,较高的不确定性。作为一些其他的例子:链信息(5',3',+,-,等等)可以被存储为生物信息学字符集20的字符中的注释数据值,并可以通过字体字符区别标记适当地表示;变异例如插入和/或缺失(通常,“插入缺失”)可以被存储为生物信息学字符集20的字符中的注释数据值,并可以通过字体字符区别标记适当地表示,例如,垂直,水平或倾斜的删除线标记(用于删除)或克拉标记(即“^”)(用于插入,沿袭文本编辑器的标记惯例),或通过使用挖空或填充笔画字体字符,等等。
为了在显示使用生物信息学字体40的由生物信息学字符集20表示的碱基或肽序列时同时传送不同类型的信息,指示不同类型的注释数据的各种区别标记可以被结合到生物信息学字体40。例如,(再次参考表1的生物信息学字符集20的实施例)字符0000001000110000bin(0230hex)表示碱基质量值的范围为51-60、是外显子的一部分的腺嘌呤碱基。另一方面,字符1000001000110000bin(8230hex)表示碱基质量值的范围为51-60、是外显子的一部分、同时也是疾病标志的一部分的腺嘌呤碱基。用于后一个字符的映射的字体字符与用于前一个字符的映射的字体字符适当地仅仅不同于增加指示是疾病标志的一部分的属性的区别标记。此外,碱基质量值可以使用合适的如在图2中示出的区别标记所描绘。如果若干不同类型的碱基或肽的属性或特征被类似地编码,那么生物信息学字体40中的字体字符的数量可以是相当大的。对于一个字节的字符,可能有多达256个不同的字体字符,而对于2字节字符可能有多达65,536个字体字符。
再参照表1的例子中,各种注释(例外的碱基质量值)由用于每个注释的单个位来表示。因此,例如,位b10的值为1表示碱基是内含子的一部分,而位b10的值为0表示碱基不是内含子的一部分。然而,这种表示方法有一个潜在的缺点,如果位b10具有零值,其可能是不明确的它是否是(1)一个肯定的表示,该碱基不是内含子的一部分,或(2)还没有(尚未)确定该碱基是否是内含子的一部分的指示。这是因为,格式转换模块16为注释位分配的默认值为零(不同于那些指示碱基质量值的)。
参照表2,生物信息学字符集20的另一个说明性的实施例被示出,其通过为注释的未知的值提供一个独特的值来克服这种不确定性。在这个实施例中,内含子的注释是由2个位b7b6表示的。值01bin表示碱基不是内含子的一部分;值10bin表示碱基是内含子的一部分;以及值1lbin表示没有(尚未)知晓该碱基是否是内含子的一部分。
表2的例子示出了生物信息学字符集20的特定实施例的一些其他预期的特征。表2的例子采用单字节字符(而表1的例子中采用了两个字节的字符)。表2的例子还使用4个位b3b2b1b0来表示碱基,从而使得不明确的碱基得到更详细的表示。该碱基的表示也使用将特定的位与特定碱基相关联的编码方案。因此,位b3与胸腺嘧啶相关联;位b2与鸟嘌呤相关联;位b1与胞嘧啶相关联;以及位b0与腺嘌呤相关联。有了这个编码方案,明确的碱基是由四个位b3b2b1b0的单个位有为1的值来表示。两种可能的碱基之间的不确定是由四个位b3b2b1b0的两个位有为1的值来表示,从而识别了碱基的两种可能性。完全不确定的端点情况由所有四个位b3b2b1b0有为1的值来表示,指示碱基可以是腺嘌呤,胞嘧啶,鸟嘌呤或胸腺嘧啶的任何一个。这种编码方案也使得不确定的碱基快速匹配到模式。例如,通过使用按位掩码04hex,字符c可以通过比较cmp[c&04hex,04hex]与由04hex表示的碱基鸟嘌呤相比较(其中cmp[...]是比较操作的伪代码)。如果字符c毫不含糊地编码为鸟嘌呤,这种比较将产生一个匹配,如果该字符c是模糊的但编码鸟嘌呤作为一个可能的值,也将产生一个匹配,在这两种情况下c&04hex=04hex。
表2
参照图6和图7并继续参照表2的例子,在一些实施例中,生物信息学字体40的字体字符不直接存储。相反的,碱基或肽字母(或三个字母)的位图表示被存储,与表示各种用于不同类型的注释的区别标记的位图一起。然后用于描绘生物信息学字符集20的特定字符的字体字符可以通过使用逻辑“或”运算将这些构成位图组合起来而被构造。在图6中最左边的术语显示了编码4个碱基的位图(任何不确定的碱基由符号“?”表示)。中间的术语显示了表示用于cds开始(以实框表示)和cds结束(由虚线框表示)的区别标记的位图。最右边的术语显示了表示用于内含子的区别标记的位图(表示为交叉通过代表碱基的字母的斜线)。图6中的加号(+)符号表示位图的逻辑或(具有一个逻辑表:黑点+黑点=黑点;黑点+白点=黑点;白点+黑点=黑点;白点+白点=白点)。图7示出了使用图6中所示的生物信息学字体40的一个实施例描绘的部分碱基序列,包括cds开始区域70,内含子区域72和cds结束区域74。区别标记“/”指示属于内含子的碱基,直观地通知审阅者该内含子不包括在翻译后蛋白质中。
参考图8,公开的序列比对/分析模块14的另一优点在于,在描绘基因组或蛋白质组序列时,它在计算上是便利的以使用户能够选择哪些注释来查看。注释类型的这种选择性的描述可能是有用的,使得审阅者专注于序列的某一方面。图8的例子采用了表1的生物信息学字符集20的实施例。图8示出了在显示设备52(例如,图1的计算机50的显示器)上显示的基因组序列的一部分。该显示还示出了用户对话框选择选项,包括复选框80,通过它审阅者选择是否显示碱基质量值(例如,使用如在图2中所示的区别标记),复选框82,通过它审阅者选择是否显示功能区域(例如,使用如在图4中所示的显示基因组区域的区别标记),以及复选框84,通过它审阅者选择是否显示变异(例如,使用如在图4中所示的区别标记来突出疾病标志)。用户适当地使用指针输入设备(例如,鼠标、轨迹球、触控板等等)、键盘(例如,使用tab键循环各选项并按下enter键来触发选中的复选框)等等来选中或取消各个复选框80、82、84。在图8中,用户已经通过复选框82选择了只显示功能区域。
如在图8中所示的显示选项使用公开的生物信息学字符集20和生物信息学40字体在输入字符串到文本显示程序(例如,由平台提供来显示ascii或unicode文本)之前通过使用位掩码来移除不要被显示的注释是容易实现的。例如,考虑表1的例子,一个只显示功能区域而不显示碱基质量值或疾病标志的合适的位掩码是mdisplay=011111110000111bin(7f87hex),它将碱基质量值(位b6b5b4b3)设置为0000bin,并将疾病标记注释(位b15)设置为0。这种方法假定生物信息学字体40不带区别标记显示最低基本质量值(即0000bin),如图2的说明性的方法的情况。注意,该掩码在输入到文本显示程序之前被应用,但掩码输出不被用来更新存储的序列30。因此,在数据文件30中,碱基质量值和疾病标记注释保持不变,因此,审阅者可以在任何时候选择更新显示选项。例如,审阅者可能会分别通过取消勾选框82和选中框84来选择关闭功能区域标记和打开变异标记。更新后的显示会通过更新掩码mdisplay=1000000000000111bin(8007hex)来容易地实现,它将碱基质量值(位b6b5b4b3)设置为0000bin,并将功能区域注释(位b14b13b12b11b10b9b8b7)设置为0。
本说明性的实施例已采用基因组数据,并且更具体地,使用四种碱基腺嘌呤,胞嘧啶,鸟嘌呤和胸腺嘧啶的dna数据。然而,所公开的采用生物信息学字符集20和生物信息学字体40的方法很容易用于其它基因组数据,如通过用尿嘧啶取代胸腺嘧啶来用于rna序列。
参考图9,所公开的采用生物信息学字符集20和生物信息学字体40的方法也很容易用于蛋白质组数据,通过采用适当数量的位来表示氨基酸(或肽)。蛋白质从一组20个氨基酸构造而来,其可以使用具有32个可能的值的5位来表示。因此5位也是足以表示任何额外的可能感兴趣的肽,如硒代半胱氨酸(通常用字母“u”或“u”或三个字母代码“sec”来表示)和吡咯赖氨酸(“o”或“o”或“pyl”),它可以通过覆盖cds结束密码子,和/或表示不确定的氨基酸(其中对大部分蛋白质组测序技术通常只有半打或更少的成对的不确定可能会出现)而被合并。图9示出通过使用逐位或操作将用于氨基酸或肽的位图和用于一个或多个区别标记的一个或多个位图相结合来构造表示各种说明性的包括各种注释数据的肽或氨基酸的生物信息学字体40的字体字符。在图9中,最上面的部分100指定20个氨基酸的合适的单字母编码。图9的中间部分102指定注释数据的各种组合,其可与蛋白质序列的肽相关联。在图9的示例性例子中,可以被注释到肽的属性包括:疏水性-由一个上标星号(*)表示;极性-由前面的“∞”区别标记表示;小的-由下标“o”;微小的-由一个下标点表示;芳香-由下标“房子”表示
应当指出的是,在一般情况下,生物信息学字符集20和相关的生物信息学字体40将特定于基因组数据,或蛋白质组数据。换句话说,给定的生物信息学字符集20和相关的生物信息学字体40的实施例被设计来表示基因组数据,或蛋白质组数据,但通常不是基因组和蛋白质组数据两者。(可预期的,字符集和相关联的字体的“组合”的实施例能够表示基因组和蛋白质组数据两者,但通常使用特定的基因组或蛋白质组字符集和字体使得更有效的生物信息学字符集和更直观的生物信息学字体的构建成为可能)。
还应当注意的是,一个给定的生物信息学字符集20可以具有两个以上不同的生物信息学字体40与其相关联。通过说明性类推的方式,以相同的方式,该ascii字符集的字符可以通过不同的字体(例如,timesnewroman字体,arial字体,等等)来表示,不同的生物信息学字体40可以被用来表示单个生物信息学字符集20。然后,用户可以通过选择用户的首选生物信息学字体40来简单地选择他或她首选的基因组或蛋白质序列的表达,以同样的方式,读者可以选择使用timesnewroman字体、arial字体或读者可能喜欢的其他任何可用的ascii字体来显示或打印英文文本。
公开的采用生物信息学字符集20和生物信息学字体40的方法进一步被容易地用来适应附加的或其他注释类型。在基因组序列的情况下,感兴趣的注释包括(但不限于):甲基化,乙酰化,cds起始和终止,外显子起始和终止,内含子起始和终止,启动子,增强子,tf,假基因,sts,d-环,v-环,mirna,pirna,ncrna,重复(线性,正弦等),gap,疾病的特定签名,等等。在蛋白质序列的情况下,感兴趣的注释类型可以包括变体,如单核苷酸变异(snv)或取代,三级结构信息,等等。
在表1和2图示的例子中,只有一个变异注释被示出(表1的例子中的疾病标志位b15)。然而,可以理解的是,更多和/或不同的变异注释可以被包括在该生物信息学字符集20中。例如,不同的注释可以被提供用于不同的疾病标志,或注释可以被提供用于其它类型的变异,诸如指示祖先谱系的变异或与特定表型性状相关的变异。
本申请描述了一个或多个优选实施例。修改和变更可以在阅读并理解了前述的详细说明的情况下发生。意图是,该申请解释为包括所有这些修改和变更,只要它们在所附权利要求或其等同物的范围之内。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种新型的15种人乳头瘤病毒亚型分型试剂盒和方法与流程
- 一种与水稻稻瘟病抗性基因紧密连锁的分子标记、引物及其应用的制造方法与工艺
- 一种无创高通量甲基化结肠癌诊断、研究和治疗方法与流程
- 用于检测CDKN2B基因变异的试剂盒及CDKN2B基因变异率的定量检测方法与流程
- 用于基因甲基化检测的引物和探针、采样方法、试剂盒与流程
- 一种癌症相关基因突变高灵敏度检测方法和试剂盒与流程
- 基于高通量测序法的人类多基因突变检测试剂盒的制造方法与工艺
- 一种用于检测德尔卑沙门氏菌的靶基因、特异性引物对及检测方法和试剂盒与流程
- 一种检测测序平台索引序列污染的方法及引物与流程
- 无物种限制的真核生物同时进行基因敲除和基因过表达的制造方法与工艺