一种基于协同矩阵分解的药物靶点相互作用关系预测方法与流程
本发明涉及药物靶点相互作用关系预测技术领域,尤其涉及一种基于协同矩阵分解的药物靶点相互作用关系预测方法。
背景技术:
药物靶点是药物与人体作用的结合位点,包括基因、受体、酶、离子通道、转运体、核酸等,通过药物与位点的结合影响生物学事件的改变,从而实现药物的治疗效果。药物-靶点相互作用(drug-targetinteractions,dti)的鉴定是现代药物发现和开发的基础。药物-靶点相互作用预测对药物发现、药物副作用预测、药物重定位,以及发现与现有药物相互作用的新靶点的过程有重要的作用。传统用于鉴定新的dti的生物化学实验方法所需设备极其昂贵且耗费时间较长。随着公开的化学生物数据库的出现,近些年出现的药物靶点数据库有phid、therapeutictargetdatabase(ttd)、drugbank、bindingdb、pharmgkb、chembl等,这些数据资源为dti的预测提供了基础。近年来,结合计算机技术可以快速低成本地识别潜在的dti。
目前的研究方法中,药物-靶点相互作用关系预测可以分为三种:基于配体的方法、基于分子对接的方法和基于化学基因组学的方法。基于配体的方法假设相似的药物具有相似的性质,也会结合相似的靶点蛋白,通过使用配体之间的相似性来预测新的dti。基于分子对接方法通过利用药物和靶点的3d结构预测药物和靶标的相互作用,广泛用于生物学。化学基因组学是一门通过结合基因组学与化学资源来研究活性化合物与细胞内靶点的潜在关系,并应用于药物和靶点发现的新兴学科。基于化学基因组学的方法可以在短时间内进行大规模预测,为实验筛选提供候选药物或者靶点,根据采用的方法又可分为三类:基于分类的方法、基于网络扩散的方法和基于矩阵分解的方法。基于分类的方法一般使用药物-药物和靶点-靶点之间的相似性、药物和靶点的结构特征,利用机器学习的方法预测药物-靶点相互作用关系的有无;基于网络扩散的方法将药物和靶点看作节点,药物-靶点之间的关系看作边,增加药物-药物、靶点-靶点之间的相似性关系等构建网络,在网络上使用随机游走等网络传播方法预测未知的药物-靶点相互作用关系。
基于矩阵分解的方法利用矩阵分解将药物靶点关联关系矩阵分解为两个低秩矩阵,对应于药物和靶点的特征空间。splcmf模型[l.y.xia,z.y.yang,h.zhang,etal.improvedpredictionofdrug-targetinteractionsusingself-pacedlearningwithcollaborativematrixfactorization[j].journalofchemicalinformationandmodeling,2019]将自步学习集成到基于协同过滤的矩阵分解中,整合药物相似度网络、靶点相似度网络和已知dti网络到正则最小二乘中,进一步提高模型的预测能力,并且在存在重噪声和丢失数据的情况下可以有效地避免不良的局部最小值。
上述的药物-靶点相互作用预测取得一定进展,但仍存在以下问题:1、基于配体的方法对于拥有少数已知配体的靶点效果不好;2、基于分子对接方法存在模拟过程耗时和许多靶点的3d结构未知的问题;3、目前存在的基于矩阵分解的方法只考虑到药物和靶点的属性特征,没有考虑dti网络中药物和靶点节点的网络潜在特征,即忽略了药物和靶点的拓扑特征,不能准确的表示药物和靶点的特征,从而导致预测结果不准确。
技术实现要素:
本发明所要解决的技术问题是现有的基于矩阵分解的方法药物和靶点的表示信息有限,并且没有考虑药物或者靶点的局部几何结构信息,导致很多情况下精确度不高的问题。本发明提供了解决上述问题的一种基于协同矩阵分解的药物靶点相互作用关系预测方法,提出图正则化约束来保持药物-靶点相互作用网络中的拓扑结构信息,使得在原始的数据空间中相似的药物或者靶点,在矩阵分解重构后也保持相似的特性,有效提高药物靶点相互作用关系预测精度。
本发明通过下述技术方案实现:
一种基于协同矩阵分解的药物靶点相互作用关系预测方法,该方法包括以下步骤:
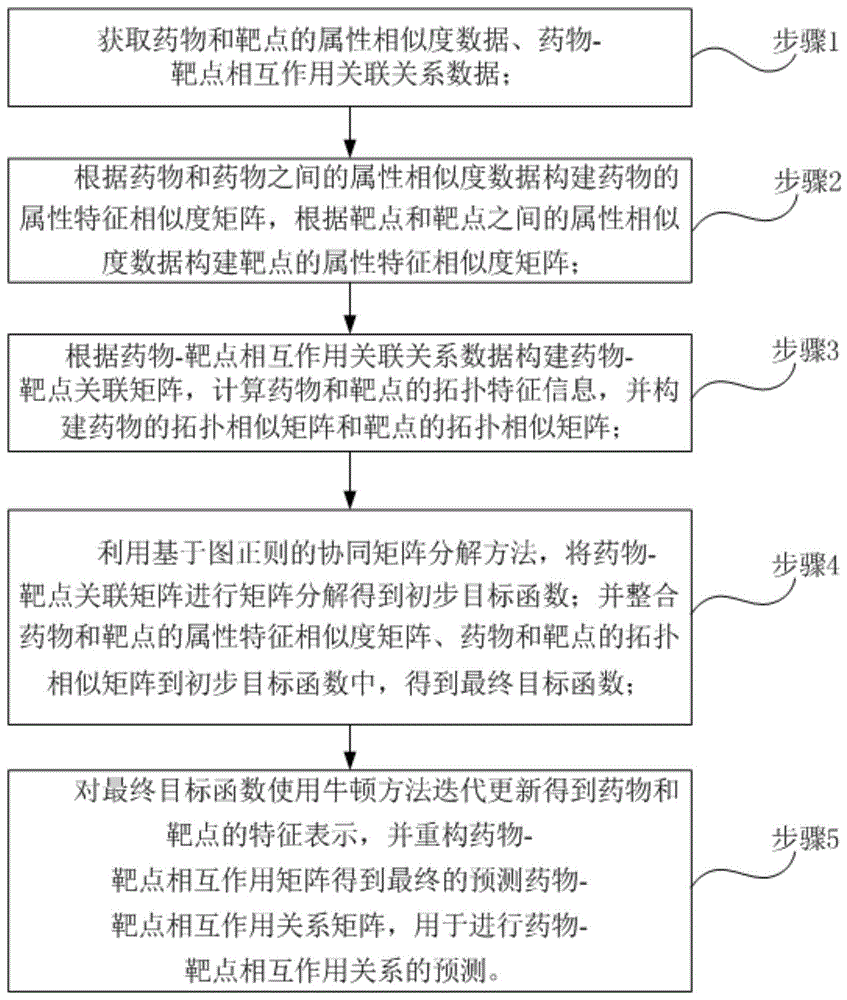
步骤1:获取药物和靶点的属性相似度数据、药物-靶点相互作用关联关系数据;
步骤2:根据药物和药物之间的属性相似度数据构建药物的属性特征相似度矩阵,根据靶点和靶点之间的属性相似度数据构建靶点的属性特征相似度矩阵;
步骤3:根据药物-靶点相互作用关联关系数据构建药物-靶点关联矩阵,计算药物和靶点的拓扑特征信息,并构建药物的拓扑相似矩阵和靶点的拓扑相似矩阵;
步骤4:利用基于图正则的协同矩阵分解方法,将药物-靶点关联矩阵进行矩阵分解得到初步目标函数;并整合药物和靶点的属性特征相似度矩阵、药物和靶点的拓扑相似矩阵到初步目标函数中,得到最终目标函数;
步骤5:对最终目标函数使用牛顿方法迭代更新得到药物和靶点的特征表示,并重构药物-靶点相互作用矩阵得到最终的预测药物-靶点相互作用关系矩阵,用于进行药物-靶点相互作用关系的预测。
工作原理是:现有的基于矩阵分解的方法药物和靶点的表示信息有限,并且没有考虑药物或者靶点的局部几何结构信息,导致很多情况下精确度不高的问题。本发明方法不仅考虑药物-药物之间和靶点-靶点之间的属性相似性,同时结合药物-药物之间和靶点-靶点之间的拓扑结构相似性;本发明方法分别计算药物-药物之间和靶点-靶点之间的属性相似性和拓扑结构相似性,基于图正则的协同矩阵分解模型,不仅可以学习到药物和靶点的属性特征,还可以学习到dti(即药物-靶点相互作用)网络中药物和靶点的拓扑特征,能够提高预测药物和靶点之间的相互作用关系的准确率。
进一步地,步骤2中根据药物和药物之间的属性相似度数据构建药物的属性特征相似度矩阵,具体地,所述药物-药物属性相似度矩阵中的两个药物之间的相似度计算公式为:
式中,|di∩dj|表示两个药物di和dj之间的最大公共子图中原子的数量,|di∪dj|表示di和dj数量之和减去di和dj之间的最大公共子图中原子的数量,即di和dj的并集;
为了便于计算机输入,将nd个药物之间的相似度全部计算出来,使用药物-药物属性相似度矩阵
进一步地,步骤2中根据靶点和靶点之间的属性相似度数据构建靶点的属性特征相似度矩阵,具体地,靶点-靶点属性相似度矩阵中的两个靶点之间的相似度计算公式为:
式中,ti和tj表示靶点列表中第i个和第j个靶点,sw(·,·)表示原始的smith–waterman分数;
将nt个靶点之间的相似度全部计算出来,使用靶点-靶点属性相似度矩阵
进一步地,由于目前仅利用药物的属性信息计算药物相似性,忽略了药物在药物-靶点网络中的拓扑信息的相似性,不能更准确的计算药物-药物的相似性。为了提取药物在药物-靶点相互作用网络中的拓扑特征,步骤3中药物和靶点的拓扑特征信息的提取均采用node2vec方法,具体地,药物拓扑特征计算包括:
(1)已知dti矩阵y,
(2)根据y矩阵构建无权值无向的网络图g=(v,e),v是节点集合,其中|v|=nd+nt,|v|表示节点的数量;e是边集合,其中
式中,z是归一化常数,πvx是节点v和x之间的转移概率,指的是节点v下一步访问节点x的概率;πvx的取值情况:
式中,dvx表示节点v和节点x之间的最短路径,参数p和q用来控制随机游走的速度和距初始节点u的距离;
(3)目标函数表达的含义是,在存在特征表达f的条件下,最大化发现节点u的网络邻居ns(u)的log概率,node2vec的目标函数为:
用f:v→rd表示节点到拓扑特征表达的映射函数,d是拓扑特征空间的维度;对于任意一个节点u∈v,
(4)使用随机梯度下降优化node2vec的目标函数,最后得到药物的d维拓扑特征,nd个药物的d维拓扑特征矩阵表示为
进一步地,和计算药物的拓扑特征相同,采用node2vec方法来计算靶点拓扑特征,得到靶点的d维拓扑特征,nt个靶点的d维拓扑特征矩阵表示为
进一步地,步骤3中构建药物和靶点的拓扑相似矩阵,计算药物-药物拓扑特征相似性、靶点-靶点拓扑特征相似性;具体地,药物-药物拓扑特征相似性计算是结合药物的拓扑特征信息,两个药物向量在拓扑特征空间下的相似度可通过余弦相似度表示;给定两个药物di和dj的拓扑特征向量表示xi和xj,使用余弦相似度计算药物-药物拓扑特征相似性:
将nd个药物之间的相似度全部计算出来,使用药物-药物拓扑特征相似度矩阵
同时:和计算药物-药物拓扑特征相似性相同,最后得到靶点-靶点的拓扑特征相似度矩阵
进一步地,步骤4详细步骤如下:
为了充分利用药物-药物和靶点-靶点的相似信息,得到药物和靶点的潜在特征表示,进行协同矩阵分解,将已知药物-靶点相互作用关系矩阵y分解成两个低秩矩阵a和b,分别对应药物和靶点的特征空间。用a和b的内积近似药物-靶点相互作用关系矩阵y:
y≈abt
式中,a和b分别是nd×k,nt×k,k是特征空间的维度。
基于已知的药物-靶点相互作用关联关系,使用最小化重构误差来估计矩阵分解中的低秩矩阵a和b,矩阵分解初步目标函数为:
式中,
整合药物-药物属性特征相似度、靶点-靶点属性特征相似度到目标函数中,得到:
cmf(协同矩阵分解)的原理是让已知药物-靶点相互作用关系y矩阵近似于a和b的内积。λl、λd、λt、λm、λn是正则化参数。属性特征相似度矩阵sd和st的矩阵分解,其过程公式表示如下:
sd≈aat
st≈bbt
pd矩阵描述了药物之间的结构相似关系,为了整合药物信息,在低维空间中保留药物图形结构,引入基于药物图形拓扑的正则化术语:
同理,pt矩阵描述了靶点之间的结构相似关系,引入基于靶点图形拓扑的正则化术语:
最终目标函数为:
式中,nd表示药物的数量,nt表示靶点的数量;w是nd和nt的权重矩阵,wij=1表示药物-靶点相互作用关系是已知的,否则表示是未知的;y表示已知药物-靶点相互作用关系;λl、λd、λt、λm、λn是正则化参数;a和b分别表矩阵分解得到的药物和靶点的特征向量;sd矩阵表示药物之间的属性特征相似度矩阵,st矩阵表示靶点之间的属性特征相似度矩阵;pd矩阵描述了药物之间的结构相似关系,pt矩阵描述了靶点之间的结构相似关系。
进一步地,步骤5中对最终目标函数使用牛顿方法迭代更新得到药物和靶点的特征表示,具体包括:
由于药物潜在特征向量ai和靶点潜在特征向量bj耦合在一起,不容易求解,因此使用牛顿方法更新ai和bj。
首先,计算l相对于ai和bj的偏导数:
然后,计算l相对于ai和bj的二阶导数:
由于矩阵
进一步地,步骤5中的dti矩阵(即药物-靶点相互作用矩阵)得到的最终的预测药物-靶点相互作用关系矩阵f为:
f=abt
式中,a和b为将已知药物-靶点相互作用关系矩阵分解成两个矩阵,a和b分别是nd×k,nt×k,k是特征空间的维度。
本发明与现有技术相比,具有如下的优点和有益效果:
1、本发明一种基于协同矩阵分解的药物靶点相互作用关系预测方法,本发明给出了一种药物-靶点预测的框架,该方法不仅考虑了药物和靶点的属性特征,还考虑了药物和靶点的拓扑结构特征,通过提出的正则项对相关性进行约束;
2、本发明一种基于协同矩阵分解的药物靶点相互作用关系预测方法,本发明基于图正则协同矩阵分解模型,提出图正则化约束来保持药物-靶点相互作用网络中的拓扑结构信息,从而提高预测药物-靶点相互作用关系的精确度。
附图说明
此处所说明的附图用来提供对本发明实施例的进一步理解,构成本申请的一部分,并不构成对本发明实施例的限定。在附图中:
图1为本发明一种基于协同矩阵分解的药物靶点相互作用关系预测方法流程图。
图2为本发明方法的基于图正则的协同矩阵分解模型图。
图3为本发明方法的基于图正则的协同矩阵分解过程图。
图4为本发明方法的属性特征相似度矩阵分解过程图。
具体实施方式
为使本发明的目的、技术方案和优点更加清楚明白,下面结合实施例和附图,对本发明作进一步的详细说明,本发明的示意性实施方式及其说明仅用于解释本发明,并不作为对本发明的限定。
实施例1
如图1至图4所示,本发明一种基于协同矩阵分解的药物靶点相互作用关系预测方法,该方法包括以下步骤:
步骤1:获取药物和靶点的属性相似度数据、药物-靶点相互作用关联关系数据;
步骤2:根据药物和药物之间的属性相似度数据构建药物的属性特征相似度矩阵,根据靶点和靶点之间的属性相似度数据构建靶点的属性特征相似度矩阵;
步骤3:根据药物-靶点相互作用关联关系数据构建药物-靶点关联矩阵,计算药物和靶点的拓扑特征信息,并构建药物的拓扑相似矩阵和靶点的拓扑相似矩阵;
步骤4:利用基于图正则的协同矩阵分解方法,将药物-靶点关联矩阵进行矩阵分解得到初步目标函数;并整合药物和靶点的属性特征相似度矩阵、药物和靶点的拓扑相似矩阵到初步目标函数中,得到最终目标函数;
步骤5:对最终目标函数使用牛顿方法迭代更新得到药物和靶点的特征表示,并重构药物-靶点相互作用矩阵得到最终的预测药物-靶点相互作用关系矩阵,用于进行药物-靶点相互作用关系的预测。
本发明技术方案使用的数据来自于yamanishi的数据集[yamanishi,y.;araki,m.;gutteridge,a.;honda,w.;kanehisa,m.predictionofdrug-targetinteractionnetworksfromtheintegrationofchemicalandgenomicspaces.bioinformatics2008,24,i232-i240.],其中根据靶点类型分为核受体(nr)、g蛋白偶联受体(gpcr)、离子通道(ic)和酶(e)四个数据集。nr数据集包括54个药物、26个靶点和90条药物-靶点相互作用关联关系;gpcr数据集包括223个药物、95个靶点和635条药物-靶点相互作用关联关系;ic数据集包括210个药物、204个靶点和1476条药物-靶点相互作用关联关系;e数据集包括445个药物、664个靶点和2926条药物-靶点相互作用关联关系;
本发明技术方案的主流程图如图1所示,主要包括以下步骤:
(1)计算药物-药物属性相似性
从yamanishi数据集中获取药物-药物属性相似度数据,yamanishi数据集中药物-药物属性相似度数据使用simcomp工具计算两个药物的相似性。simcomp工具提供基于公共子结构的全局相似性分数计算,其原理是:
给定两个药物di和dj,分别表示药物列表中第i和第j个药物的化学结构,由原子作为顶点和共价键作为边缘的2d图形。首先从yamanishi的数据集获得药物列表,即药物的名称列表;然后,在keggligand数据库中查询药物的化合物编号;最后,在simcomp工具中输入其中一个药物的化合物编号,该工具根据该化合物的2d图形计算与其他药物的最大公共子结构,最后按相似程度由高到低排列药物。
具体相似度计算方法是使用jaccard相似度进行计算两个药物的相似度:
式中,|di∩dj|表示di和dj之间的最大公共子图中原子的数量,|di∪dj|表示di和dj数量之和减去di和dj之间的最大公共子图中原子的数量,即di和dj的并集。为了便于计算机输入,将nd个药物之间的相似度全部计算出来,使用药物-药物属性相似度矩阵
(2)计算靶点-靶点属性相似性
从yamanishi数据集中获取靶点-靶点属性相似度数据,由于原始的smith–waterman分数受到靶点序列长度的影响,导致原始的smith–waterman分数不能准确衡量靶点之间的属性相似度,因此使用归一化smith–waterman分数计算两个靶点之间的相似度。归一化smith–waterman分数的计算如下:
式中,ti和tj表示靶点列表中第i个和第j个靶点,sw(·,·)表示原始的smith–waterman分数。为了便于计算机输入,将nt个靶点之间的相似度全部计算出来,使用靶点-靶点属性相似度矩阵
(3)计算药物拓扑特征
由于目前仅利用药物的属性信息计算药物相似性,忽略了药物在药物-靶点网络中的拓扑信息的相似性,不能更准确的计算药物-药物的相似性。为了提取药物在药物-靶点相互作用网络中的拓扑特征,本发明使用node2vec方法。
已知dti矩阵y,
根据y矩阵构建无权值无向的网络图g=(v,e),v是节点集合,其中|v|=nd+nt,|v|表示节点的数量。e是边集合,其中
为了获取药物的拓扑特征,在网络图g上进行二阶随机游走,给定源节点u,随机游走的固定长度l,ci表示游走过程中第i个节点,游走的起点是c0=u。节点ci由如下公式产生:
式中,z是归一化常数,πvx是节点v和x之间的转移概率,指的是节点v下一步访问节点x的概率。πvx的取值情况:
式中,dvx表示节点v和节点x之间的最短路径。参数p和q用来控制随机游走的速度和距初始节点u的距离。
node2vec的目标函数为:
用f:v→rd表示节点到拓扑特征表达的映射函数,d是拓扑特征空间的维度。对于任意一个节点u∈v,
最后得到药物的d维拓扑特征,nd个药物的d维拓扑特征矩阵表示为
(4)计算靶点拓扑特征
和计算药物的拓扑特征相同,最后得到靶点的d维拓扑特征,nt个靶点的d维拓扑特征矩阵表示为
(5)计算药物-药物拓扑特征相似性
结合药物的拓扑特征信息,两个药物向量在拓扑特征空间下的相似度可通过余弦相似度表示。给定两个药物di和dj的拓扑特征向量表示xi和xj,使用余弦相似度计算药物-药物拓扑特征相似性:
将nd个药物之间的相似度全部计算出来,使用药物-药物拓扑特征相似度矩阵
(6)计算靶点-靶点拓扑特征相似性
和计算药物-药物拓扑特征相似性相同,最后得到靶点-靶点的拓扑特征相似度矩阵
(7)基于图正则的协同矩阵分解
为了充分利用药物-药物和靶点-靶点的相似信息,得到药物和靶点的潜在特征表示,进行协同矩阵分解,将已知药物-靶点相互作用关系矩阵y分解成两个低秩矩阵a和b,分别对应药物和靶点的特征空间。如图2所示,用a和b的内积近似药物-靶点相互作用关系矩阵y:
y≈abt
式中,a和b分别是nd×k,nt×k,k是特征空间的维度。
基于已知的药物-靶点相互作用关联关系,使用最小化重构误差来估计矩阵分解中的低秩矩阵a和b,矩阵分解初步目标函数为:
式中,
整合药物-药物属性特征相似度、靶点-靶点属性特征相似度到目标函数中,得到:
cmf(协同矩阵分解)的原理是让已知药物-靶点相互作用关系y矩阵近似于a和b的内积。λl、λd、λt、λm、λn是正则化参数。属性特征相似度矩阵sd和st的矩阵分解如图4所示,其过程公式表示如下:
sd≈aat
st≈bbt
pd矩阵描述了药物之间的结构相似关系,为了整合药物信息,在低维空间中保留药物图形结构,引入基于药物图形拓扑的正则化术语:
同理,pt矩阵描述了靶点之间的结构相似关系,引入基于靶点图形拓扑的正则化术语:
最终目标函数为:
(8)优化目标函数
由于药物潜在特征向量ai和靶点潜在特征向量bj耦合在一起,不容易求解,因此使用牛顿方法更新ai和bj。
首先,计算l相对于ai和bj的偏导数:
然后,计算l相对于ai和bj的二阶导数:
由于矩阵
(9)药物-靶点相互作用关系预测
重构dti矩阵得到最终的预测药物-靶点相互作用关系矩阵:
f=abt
根据以上步骤,实施本发明方法,本发明方法不仅考虑药物-药物之间和靶点-靶点之间的属性相似性,同时结合药物-药物之间和靶点-靶点之间的拓扑结构相似性;本发明方法分别计算药物-药物之间和靶点-靶点之间的属性相似性和拓扑结构相似性,基于图正则的协同矩阵分解模型,考虑将网络的拓扑结构作为正则化项融入协同矩阵分解过程中,不仅可以学习到药物和靶点的属性特征,还可以学习到dti(即药物-靶点相互作用)网络中药物和靶点的拓扑特征,充分利用了节点属性和网络拓扑互为补充的优势,能够提高预测药物和靶点之间的相互作用关系的准确率。
现有背景技术方案通常只使用药物和靶点属性相似度矩阵进行药物靶点相互作用关系预测,这种方法可以获得节点的潜在空间表示,但由于没有考虑网络的拓扑结构信息,影响预测精度。根据“guiltbyassociation”假设,两种类似的药物往往具有共同的靶点,或者说两个具有类似拓扑结构的节点,往往体现着相似的功能。因此本发明将网络的拓扑结构也融入到模型中。这样,即使两个药物的属性不同,但如果它们在网络中共享相似的连接,它们也可能拥有共同的靶点;反之,药物和靶点之间即使缺乏连接,但如果两个药物有相似的属性,则也可能拥有共同的靶点。双方信息互补,从而提高药物和靶点之间的相互作用关系预测精度。
以上所述的具体实施方式,对本发明的目的、技术方案和有益效果进行了进一步详细说明,所应理解的是,以上所述仅为本发明的具体实施方式而已,并不用于限定本发明的保护范围,凡在本发明的精神和原则之内,所做的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!