鱼鳔来源肝素类粘多糖在制备血管生成抑制剂中的应用的制作方法
本发明涉及医药技术领域,具体涉及鱼鳔来源肝素类粘多糖在制备血管生成抑制剂中的应用。
背景技术:
鱼鳔,又名鳔胶,是鱼体腔内主司沉浮的胶质囊。鱼鳔在我国沿海地区的食用历史悠久,作为我国传统的药食两用海洋水产特色食物资源,素有“鱼中人参”之誉,是传统中药材(《本草纲目》、《全国中草药汇编》)。鱼鳔作为中药材,具有独特的滋补作用和药用价值,具有补肾益精,滋养筋脉,止血,散瘀,消肿等功效,然而其抑制抗血管生成的用途尚未见相关报道。
技术实现要素:
鉴于此,本发明的目的在于提供鱼鳔来源肝素类粘多糖在制备血管生成抑制剂中的应用,鱼鳔来源肝素类粘多糖对血管生成的抑制作用强。
为了实现上述发明目的,本发明提供以下技术方案:
本发明提供了鱼鳔来源肝素类粘多糖在制备血管生成抑制剂中的应用,所述鱼鳔来源肝素类粘多糖结构单元为α-δglcua-[1→3]-galnac-4s。
优选的,所述鱼鳔来源肝素类粘多糖的制备方法,包括以下步骤:
将鱼鳔干粉和水混合,得到鱼鳔粉悬浮液;
将所述鱼鳔粉悬浮液、氯化钠和蛋白酶混合,进行酶解,得到酶解液;
将所述酶解液灭酶后离心分离,得到上清液;
将所述上清液依次进行大孔阴离子交换树脂吸附和氯化钠水溶液洗脱,得到洗脱液;
将所述洗脱液进行沉淀后干燥,得到鱼鳔来源肝素类粘多糖。
优选的,所述氯化钠的质量为所述鱼鳔干粉质量的1.2~1.8%;
所述蛋白酶的质量为所述鱼鳔干粉质量的0.5~3.0%。
优选的,所述氯化钠水溶液的浓度为0.3~1.1mol/l。
优选的,所述血管包括人脐静脉血管或鸡胚尿囊膜血管。
优选的,所述鱼鳔来源肝素类粘多糖对人脐静脉血管的有效抑制浓度为0.5~2mg/ml。
优选的,所述鱼鳔来源肝素类粘多糖对鸡胚尿囊膜血管的有效抑制浓度为100~500mg/ml。
本发明提供了鱼鳔来源肝素类粘多糖在制备血管生成抑制剂中的应用,所述鱼鳔来源肝素类粘多糖的结构单元为α-δglcua-[1→3]-galnac-4s。在本发明中,鱼鳔来源肝素类粘多糖(简写为hsb)对于血管生成的抑制作用强。如本发明实施例结果所示,400mg/l鱼鳔来源肝素类粘多糖对人脐静脉血管内皮细胞生长的抑制率可达90.3%;1mg/ml鱼鳔来源肝素类粘多糖对鸡胚尿囊膜血管生成的抑制率为77.15%。
附图说明
图1为hsb的紫外光谱光谱图;
图2为hsb的高效凝胶色谱图;
图3为混合单糖标准品的高效液相色谱图;
图4为hsb中单糖衍生物的高效液相色谱图;
图5为水解时间对于hsb水解样品中各组分的峰面积比值的影响图;
图6为hsb的傅里叶红外光谱图;
图7为a、c、d和e型的硫酸软骨素标准品和hsb完全降解产物的ms/ms谱图;
图8为hsb中硫酸软骨素碎片基本组成图;
图9为hsb的1h谱图;
图10为hsb的13c谱图;
图11为hsb的hsqc谱图;
图12为hsb的hmbc谱图;
图13为hsb对人脐静脉血管内皮细胞生长的抑制效果图;
图14为鸡胚尿囊膜血管实物图,其中,a后续经hsb处理,b后续经pbs处理;
图15为hsb和pbs对鸡胚尿囊膜血管处理后的实物图,其中,a为hsb处理,b为pbs处理。
具体实施方式
本发明提供了鱼鳔来源肝素类粘多糖在制备血管生成抑制剂中的应用,所述鱼鳔来源肝素类粘多糖的结构单元为α-δglcua-[1→3]-galnac-4s。
在本发明中,所述鱼鳔来源肝素类粘多糖的制备方法,优选包括以下步骤:
将鱼鳔干粉和水混合,得到鱼鳔粉悬浮液;
将所述鱼鳔粉悬浮液、氯化钠和蛋白酶混合,进行酶解,得到酶解液;
将所述酶解液灭酶后离心分离,得到上清液;
将所述上清液依次进行大孔阴离子交换树脂吸附和氯化钠水溶液洗脱,得到洗脱液;
将所述洗脱液进行沉淀后干燥,得到鱼鳔来源肝素类粘多糖。
本发明将鱼鳔干粉和水混合,得到鱼鳔粉悬浮液。在本发明中,所述鱼鳔干粉的粒度优选为150~300μm,更优选为200~250μm。在本发明中,所述鱼鳔干粉优选为将鱼鳔干燥后粉碎得到。在本发明中,所述干燥的温度优选为40~60℃,更优选为50℃;本发明对于所述干燥的时间没有特殊限定,干燥至恒重即可。本发明对于所述粉碎的方式没有特殊限定,能够得到上述技术方案所述粒度的鱼鳔干粉即可。
在本发明中,所述鱼鳔干粉和水的质量比优选为1:(20~35),更优选为1:(25~30)。
得到鱼鳔粉悬浮液后,本发明将所述鱼鳔粉悬浮液、氯化钠和蛋白酶混合,进行酶解,得到酶解液。
在本发明中,所述氯化钠的质量优选为所述鱼鳔干粉质量的1.2~1.8%,更优选为1.4~1.6%,最优选为1.5%。本发明加入上述比例的氯化钠可以提高鱼鳔中蛋白质的溶解度,提高酶解效率,促进多糖从糖蛋白中分离。
在本发明中,所述蛋白酶的质量优选为所述鱼鳔干粉质量的0.5~3.0%,更优选为1~2.5%,最优选为1.5~2.0%。
本发明对于所述蛋白酶的种类没有特殊限定,采用本领域技术人员熟知的蛋白酶即可,在本发明的实施例中,所述蛋白酶优选为2709碱性蛋白酶。
在本发明中,所述酶解的温度优选为45~60℃,更优选为45~55℃,最优选为50℃;时间优选18~20h,更优选为19h;ph值优选为7.5~9,更优选为8~8.5;本发明对于所述ph值调节采用的试剂没有特殊限定,采用本领域技术人员熟知的碱即可,具体如氢氧化钠或氢氧化钾。在本发明中,所述酶解过程中,鱼鳔中的蛋白被酶解,释放出类肝素。
得到酶解液后,本发明将所述酶解液灭酶后离心分离,得到上清液。在本发明中,所述灭酶优选为高温灭酶;所述高温灭酶的温度优选为90~110℃,更优选为100℃;在本发明的实施例中,所述灭酶的温度优选由沸水浴提供;所述灭酶的时间优选为8~12min,更优选为10min。
所述灭酶后,本发明优选还包括将所述灭酶体系冷却至室温,本发明对于所述冷却的方式没有特殊限定,采用本领域技术人员熟知的冷却方式即可。
在本发明中,所述离心分离的温度优选为室温;速度优选为7000~9000r/min,更优选为8000r/min;时间优选为15~25min,更优选为20min。
得到上清液后,本发明将所述上清液依次进行大孔阴离子交换树脂吸附和氯化钠水溶液洗脱,将所得洗脱液进行沉淀后干燥,得到鱼鳔来源肝素类粘多糖。
在本发明中,所述大孔阴离子交换树脂的孔径优选为600~800μm,更优选为650~750μm,最优选为700μm;所述大孔阴离子交换树脂优选包括fpa98cl、d218、d204、d208、d254、d301型大孔阴离子交换树脂。
在本发明中,所述大孔阴离子交换树脂的重复利用率高,使用过的大孔阴离子交换树脂的处理方法,包括以下步骤:使用过的大孔阴离子交换树脂依次进行水浸泡、再生液处理和水洗。在本发明中,所述水浸泡的温度优选为室温;时间优选为10~14h,更优选为12h;所述水浸泡的过程中大孔阴离子交换树脂充分溶胀。在本发明中,所述再生液优选包括以下组分:氯化钠8~12wt%、氢氧化钠0.3~0.5wt%和余量水,所述氯化钠的含量优选为10wt%,所述氢氧化钠的含量优选为0.4wt%;所述溶胀后的大孔阴离子交换树脂和再生液的体积比优选为1:(3~5),更优选为1:4。在本发明中,所述再生液处理的时间优选为1~3h,更优选为2h;所述再生液处理过程能够将使用过的大孔阴离子交换树脂所吸附的离子和其他杂质洗脱除去,恢复至原来的组成和性能。在本发明中,所述水洗优选为蒸馏水洗;本发明对于所述水洗的次数没有特殊限定,水洗至流出液为中性即可;所述水洗的目的是除去再生液。
在本发明中,所述吸附的方式优选为动态吸附;所述吸附的温度优选为40~50℃,更优选为45℃。在本发明中,所述上清液在所述大孔阴离子交换树脂层析柱中的流速优选为0.5~2倍柱床体积/h,更优选为1~1.5倍柱床体积/h。本发明对于所述层析柱的规格没有特殊限定,采用本领域技术人员熟知的层析柱规格即可,在本发明的实施例中,所述层析柱的规格优选为0.28cm×100cm。在本发明中,所述上清液的上样量优选为3~8倍柱床体积,更优选为4~6倍柱床体积。在本发明中,所述吸附过程中,上清液通过大孔阴离子交换树脂进行选择性动态吸附,能够分离出部分杂蛋白及核酸。
所述吸附后,本发明优选还包括将所述吸附后的预处理大孔阴离子交换树脂层析柱进行水洗。本发明对于所述水洗的时间数没有特殊限定,水洗至流出液为无色透明溶液为止。
在本发明中,所述氯化钠水溶液的浓度优选为0.1~1.5mol/l。在本发明中,所述洗脱优选为梯度洗脱,具体的为依次采用浓度为0.3mol/l、0.5mol/l、0.9mol/l、1.1mol/l氯化钠水溶液进行梯度洗脱。在本发明中,所述梯度洗脱过程中收集1.1mol/l氯化钠的洗脱液,利用阿利新蓝法追踪管中类肝素含量。
得到洗脱液后,本发明将所述洗脱液沉淀后干燥,得到鱼鳔来源肝素类粘多糖。在本发明中,所述沉淀采用的沉淀试剂优选为无水乙醇,所述无水乙醇和洗脱液的体积比优选为(0.8~1.5):1,更优选为1:1;所述沉淀的时间优选为10~14h,更优选为12h。
在本发明中,所述沉淀后本发明优选还包括将所述沉淀的体系进行离心分离,将所得固体产物进行无水乙醇洗涤和透析袋脱盐。在本发明中,所述离心分离的速度优选为3500~4500r/min,更优选为4000r/min;时间优选为4~6min,更优选为5min。在本发明中,所述无水乙醇洗涤的次数优选为2~3次。在本发明中,所述透析袋的截留分子量优选为1000~3000da,更优选为2000da。本发明对于所述透析袋脱盐的操作没特殊限定,能够将体系中的氯化钠除去即可。
在本发明中,所述干燥的方式优选为冷冻干燥,所述冷冻干燥优选在冷冻干燥机中进行;本发明对于所述冷冻干燥的温度和时间没有特殊限定,冷冻干燥至恒重即可。
在本发明中,所述血管优选包括人脐静脉血管或鸡胚尿囊膜血管。在本发明中,所述鱼鳔来源肝素类粘多糖对人脐静脉血管的有效抑制浓度优选为0.5~2mg/ml,更优选为1~1.5mg/ml。在本发明中,所述鱼鳔来源肝素类粘多糖对鸡胚尿囊膜血管的有效抑制浓度优选为100~500mg/ml,更优选为300~400mg/ml。
下面将结合本发明中的实施例,对本发明中的技术方案进行清楚、完整地描述。显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
实施例1
将鱼鳔在50℃的烘箱中干燥至恒重,粉碎,得到粒度为150~300μm的鱼鳔干粉;将所述鱼鳔干粉与蒸馏水按照质量比为1:20混合,得到鱼鳔粉悬浮液。
将所述400ml鱼鳔粉悬浮液、6g氯化钠和8g2709碱性蛋白酶混合,在50℃下酶解20h,得到酶解液。
将所述酶解液置于沸水浴中灭酶10min,冷却至室温后在8000r/min下离心分离20min,得到上清液。
将罗门哈斯fpa98cl树脂用蒸馏水浸泡12h,加入再生液处理2h,用蒸馏水洗至中性;得到预处理fpa98cl树脂,置于层析柱(0.28cm×100cm)中,得到预处理fpa98cl树脂层析柱;其中,再生液组成为氯化钠10wt%、氢氧化钠0.4wt%和余量水;再生液和罗门哈斯fpa98cl树脂体积之比为5:1。
将所述上清液以1.5倍柱床体积/h的流速流入预处理fpa98cl树脂层析柱中,在45℃下动态吸附,蒸馏水冲洗层析柱至流出液为无色透明为止,然后依次采用浓度分别为0.3mol/l、0.5mol/l、0.9mol/l和1.1mol/l的氯化钠水溶液进行梯度洗脱,分别收集0.3mol/l、0.5mol/l、0.9mol/l和1.1mol/l的氯化钠水溶液进行洗脱得到的洗脱液,依次记为洗脱液f1、洗脱液f2、洗脱液f3和洗脱液f4;
分别在所述洗脱液f1~f4中加入无水乙醇进行沉淀,静置12h后在4000r/min离心分离5min,将所得固体产物无水乙醇洗涤2次,经3000kda透析袋脱盐,置于冷冻干燥机中冷冻干燥至恒重,依次得到洗脱组分f1、洗脱组分f2、洗脱组分f3和鱼鳔来源肝素类粘多糖f4(鱼鳔来源肝素类粘多糖f4简写为hbs)。
(1)鱼鳔来源肝素类粘多糖基本成分测定
鱼鳔来源肝素类粘多糖含量测定:采用阿利新蓝法,以肝素为标准品;
蛋白质含量测定:采用福林-酚法,以牛血清蛋白为标准品;
糖醛酸含量测定:采用咔唑-硫酸法,以葡萄糖醛酸为标准品;
氨基己糖含量测定:采用wagner法,以葡萄糖胺为标准品;
硫酸基含量测定:采用bacl2-gel比浊法,以硫酸钾为标准品。
洗脱液f1~f4中鱼鳔来源肝素类粘多糖的收率按照式(1)计算,鱼鳔来源肝素类粘多糖中各组分的测试指标的含量按照式(2)计算,鱼鳔来源肝素类粘多糖基本成分如表1所示。
式(1)中:md1为鱼鳔来源肝素类粘多糖的质量,单位为g;md2为鱼鳔干粉的质量,单位为g。
式(2)中,c为各组分的测试指标的含量,单位为mg/ml;md1为鱼鳔来源肝素类粘多糖的质量,单位为mg;v为洗脱液体积,单位为ml。
表1各洗脱液中鱼鳔来源肝素类粘多糖的得率和鱼鳔来源肝素类粘多糖中各组分的含量
表中,nd表示未检测出。
由表1可知,从洗脱液f1到洗脱液f4,类肝素、糖醛酸和氨基己糖含量逐渐升高,蛋白质含量递减,说明上述方法能将类肝素较好的分离出来。洗脱液f4中类肝素的得率最高,为(2.21±0.03)mg/g,f2次之;f1中蛋白质含量最高,类肝素的得率和含量都很低,且检测不到糖醛酸和氨基己糖含量,说明f1可能为中性糖和蛋白质的混合物。f4的硫酸基含量最高,为(12.29±2.20)%,f3次之,其他组分未检测到硫酸基;硫酸基含量是提示类肝素活性的一项重要指标,一定范围内硫酸基的含量越高,生物活性越强。
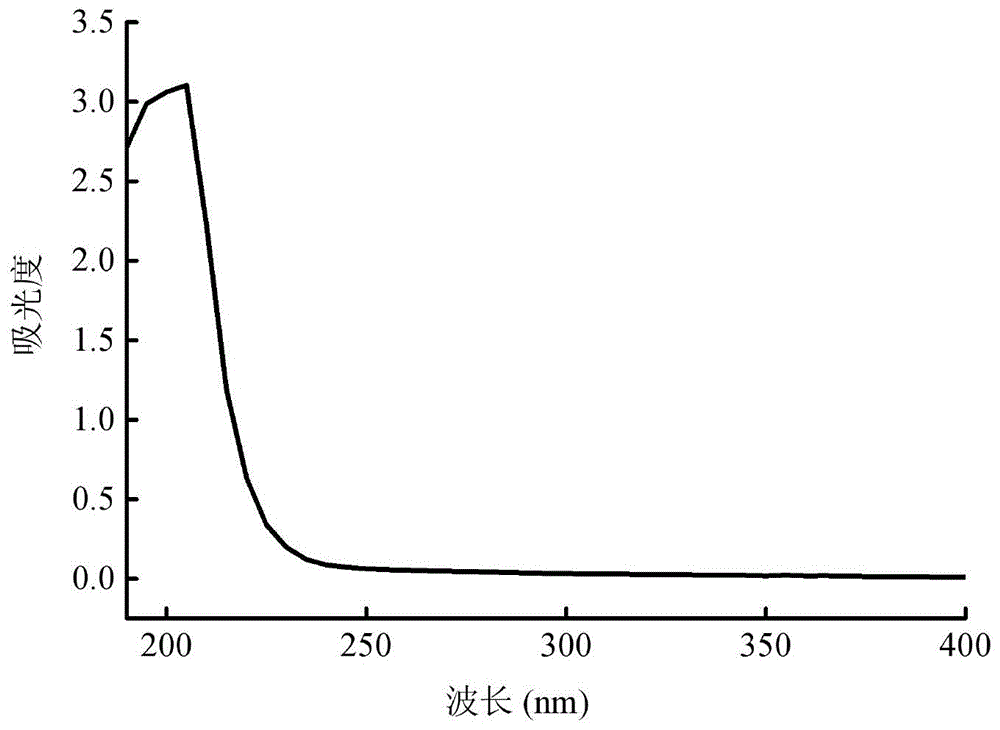
(2)hsb的紫外光谱光谱分析
将hsb用蒸馏水配成1mg/ml的hbs溶液,以蒸馏水为调零管,采用agilentcary60紫外可见分光光度计测定hbs溶液的紫外光谱吸收谱图,结果如图1所示。
由图1可知,实施例1制备的鱼鳔来源肝素类粘多糖在204nm处有糖基末端吸收外,在其他紫外区无明显吸收峰;结合表1中蛋白质含量仅为(0.19±0.07)%,表明本发明制备的鱼鳔来源肝素类粘多糖中核酸、蛋白质等杂质含量低,纯度高。
(3)hsb的高效凝胶色谱分析
采用高效凝胶色谱(hpgpc)测定hsb的纯度和分子量,测试条件:色谱柱为watersultrahydrogelcolumn500(7.8mm×300mm);柱温为35℃;流动相为0.2mol/l硫酸钠,流速为0.6ml/min;检测器为agilent1200示差检测器;进样体积为10μl。
葡聚糖标准品(10000u、25000u、50000u、80000u、150000u、270000u、410000u)和hsb分别用0.2mol/l硫酸钠配置成5mg/ml的溶液,经0.22μm的针式滤膜过滤后进样,记录葡聚糖标准品和hsb样品的保留时间进行数据处理,测试结果如图2所示。由图2可知,hsb的高效凝胶色谱图只出现一个峰,且峰形较对称,表明hsb的质量分布相对均一,纯度较高。高效凝胶色谱与示差折光检测器联用测得葡聚糖的分子量的对数(y)与保留时间(x)的回归方程为:y=-0.3192+9.429(r2=0.9966),计算得出hsb的分子质量为84033u,一般类肝素的分子质量在5000~40000u范围之间,相比之下,本发明制备的鱼鳔来源肝素类粘多糖的分子质量较大。
(4)hsb中单糖组成分析
称取3.0mghsb于15ml安倍瓶中,加入9ml1.5mol/l的三氟乙酸,置于110℃烘箱中分别水解2、4、6、8、12、16、24和28h,将得到的水解液用氮气吹干,吹干物用超纯水溶解,得到不同水解时间下的hsb水解样品;
采用单糖标准品,利用pmp衍生-高效液相反向色谱法(参见:wangq,zhaox,puj,etal.influencesofacidicreactionandhydrolyticconditionsonmonosaccharidecompositionanalysisofacidic,neutralandbasicpolysaccharides[j].carbohydratepolymers,2016,143:296-300.)测试混合单糖标准品、hsb和hsb水解样品中各组分的含量,测试结果如图3~5所示,其中,图3为混合单糖标准品的高效液相色谱图,图4为hsb中单糖衍生物的高效液相色谱图,图5为水解时间对于hsb水解样品中各组分的峰面积比值的影响图。单糖标准品为甘露糖(mannose,man)、鼠李糖(rhamnose,rha)、葡萄糖醛酸(glucuronicacid,glca),艾杜糖醛酸(iduronicacid,idoa),n-乙酰半乳糖胺(n-acetylgalactosamine,galnac)、半乳糖(galactose,gal)。pmp衍生-高效液相反向色谱法测试条件:色谱柱为agilentzorbaxeclipsexdb-c18(4.6×250mm,5μm);柱温为30℃;流动相为0.02mol/l磷酸盐缓冲液(ph=6.0)-乙腈(体积比=83:17),流速为1.0ml/min;检测器波长为245nm;进样体积为10μl。
由图3~5可知,对比单糖混标的色谱图(图3),发现hsb的高效液相色谱图(图4)出现一个峰值很高的未知峰,从图5看出,随着hsb水解时间的延长,未知峰的峰面积比例减少,葡萄糖醛酸和艾杜糖醛酸的峰面积比例也逐渐减少,n-乙酰基半乳糖胺的峰面积比例不断升高,说明未知峰是未降解寡糖片段,糖醛酸在酸溶液中不稳定,易发生脱羧反应,甚至会被强酸完全分解,因此降解时间越长糖醛酸的峰面积会减低,根据出峰时间和峰面积得出,hsb主要含葡萄糖醛酸、n-乙酰基半乳糖胺和少量的艾杜糖醛酸、半乳糖,由此得出hsb的主链可能是硫酸软骨素。
(5)hsb的傅里叶红外光谱分析
将3mghsb在红外灯下烘烤2h,然后放到玛瑙研钵中,加入300mg以同样方式处理好的氯化钾混合均匀,研磨至颗粒小于2.5μm,放入压片机中加压制成半透明的小薄片,再将其放入傅里叶红外光谱扫描仪中进行光谱扫描,扫描范围为4000~400cm-1,结果如图6所示,其中,cs-0.4表示cs的透光率减去0.4,是为了将两条曲线区分开来。由图6可知,hsb的红外光谱图与硫酸软骨素标准品基本一致,3421cm-1处出现强而宽的峰是o-h和n-h伸缩振动,说明hsb存在分子间和分子内的氢键;2918cm-1处有吸收峰表明存在亚甲基;1635cm-1处强而稍宽的峰是乙酰氨基中的c=o对称伸缩振动产生的;1500cm-1处n-h变角振动和1419cm-1处c-n伸缩振动,表明乙酰氨基的存在;1377cm-1处的峰是-coo-的c=o对称伸缩振动产生的;1255cm-1处硫酸酯基中的c=o伸缩振动、1234cm-1处硫酸基s=o伸缩振动和852cm-1处硫酸酯基的c-o-s轴向伸缩振动表明存在硫酸酯基团;927cm-1处的吸收峰是吡喃糖环的非对称伸缩振动产生的。
根据硫酸软骨素单元中硫酸基数量和链接位置的不同,硫酸软骨素分为a、c、d、e等种类,其中,a型硫酸软骨素(csa)的硫酸基在c4,产生轴向伸缩振动峰在850cm-1附近;c型硫酸软骨素(csc)的硫酸基在c6,硫酸基处于平位,吸收峰在850cm-1附近,标准品为e型硫酸软骨素,在820cm-1和850cm-1附近均有吸收峰;hsb只在850cm-1附近有的吸收峰,表明hsb可能为硫酸软骨素a或其衍生物。
(6)hsb的质谱分析
为进一步分析hsb的硫酸软骨素组成,采用硫酸软骨素酶abc将硫酸软骨素完全裂解成不饱和二糖,再经ms/ms分析鉴定。
精密称取0.0100ghsb,加入5ml醋酸铵缓冲液(ph=7.6~8.0,含0.2u硫酸软骨素酶abc),在37℃孵育24h,沸水浴灭酶5min,在10000r/min下离心25min,所得上清液用3kda超滤离心管过滤,取滤过液冷冻干燥,得到处理hsb样品。将处理hsb样品和硫酸软骨素标准品分别用超纯水配成1μg/ml溶液用于质谱/质谱分析。
质谱条件:采用电子轰击离子源;电子能量为70ev;传输线温度为275℃;离子源温度为200℃;母离子为m/z285;激活电压为1.5v;质量扫描范围为m/z35~500。
根据硫酸基团数量及链接位置,硫酸软骨素分为δdi-0s、δdi-ua-2s、δdi-4s和δdi-6s。其中,δdi-4s是csa主要硫酸软骨素单位,δdi-6s是csc的主要硫酸软骨素单位。此外,由于具有相同硫酸基团数量的硫酸软骨素相对分子质量相同,不能通过准离子峰区分开,因此通过二级质谱的碎片离子将其区分开来。四种常见的硫酸软骨素标准品和hsb完全降解产物的ms/ms结果如表2和图7所示,hsb中硫酸软骨素碎片基本组成图如图8所示。
表2hsb中硫酸软骨素碎片基本组成图
由表2和图7~8可知,δdi-0s和δdi-ua-2s的半乳糖胺不带硫酸基,特征碎片结构是[z1-2h]-,m/z为202.09;δdi-ua-2s的硫酸基连接在葡萄糖醛酸c2的羟基上,因此具有[y2-h]-和[z2+na-h]-特征碎片结构,其m/z分别为236.99、276.98。δdi-4s和δdi-6s的区别体现在,δdi-4s在m/z282处的离子丰度小于m/z300或不存在,而δdi-6s在m/z300处的离子丰度小于m/z282或不存在。图7中,δdi-4s在m/z300.06处的离子丰度大于m/z282.05,δdi-6s在m/z282.05处的离子丰度较大,在m/z300.06处没有碎片峰;δdi-6s在m/z239.94处的特征峰,其结构为[w1-h]-,可用来判断是否存在δdi-6s。hsb完全降解产物的质谱图中:不含m/z202.09、236.99和276.98,表明hsb不含δdi-0s和δdi-ua-2s;在m/z300.06处的离子丰度大于m/z282.05,且不含δdi-6s特征峰m/z239.94,说明hsb的主要硫酸软骨素单位为δdi-4s。结合红外光谱扫描结果,确定hsb主要为csa。
(7)hsb的核磁共振分析
将hsb溶于d2o中后冻干,反复三次以置换出h2o,处理后的样品用d2o配置成30mg/ml的溶液,在常温下用700m核磁共振仪进行1h谱、13c谱、异核单量子相干谱(hsqc)和异核多键相关(hmbc)分析,结果如图9~12所示,其中,图9为1h谱图,图10为13c谱图,图11为hsqc谱图,图12为hmbc谱图。
由图9~12可知,在1h谱图中,质子信号最强的峰位于4.79ppm处,是氘代水产生的溶剂峰,除此之外,所有质子信号均位于两个光谱区;在2.0~2.1ppm之间出现了乙酰胺甲基信号,而csa和csc的乙酰胺甲基信号分别在2.04ppm、2.02ppm处;hsb乙酰胺甲基信号位于2.04ppm,说明hsb可能为csa;其他质子信号集中在3~5ppm之间,说明hsb为β-构型。在13c谱图中,174.96ppm和174.35ppm的低场信号表明存在乙酰氨基和己糖醛酸的羧基;22.45ppm的高场信号可能为乙酰胺甲基碳;其他信号集中在50-105ppm之间。此外,hsqc谱图中的(2.04ppm,22.45ppm)和hmbc谱图(2.04ppm,174.38ppm)确定22.45ppm为乙酰氨基的甲基碳信号,2.04ppm为乙酰氨基的甲基质子信号,174.38ppm为乙酰氨基的羰基碳信号。参照文献(toidat,toyodah,imanarit.high-resolutionprotonnuclearmagneticresonancestudiesonchondroitinsurfates.[j].analyticalsciences,1993,9(1):53-58.和muccia,schenettil,volpin.1hand13cnuclearmagneticresonanceidentificationandcharacterizationofcomponentsofchondroitinsulfatesofvariousorigin[j].carbohydratepolymers,2000,41(1):37-45.)对hsb的1h、13c谱进行归属,如表3所示。对1h、13c、hsqc和hmbc进行综合分析,最终得出hsb的主要单元结构式为α-δglcua-[1→3]-galnac-4s。
表3hsb的1h、13c信号归属
应用例1
mtt法测定hsb对人脐静脉血管内皮细胞生长的抑制
取对数生长期人脐静脉血管内皮细胞(购买于通派(上海)生物科技有限公司的ecv304细胞株)接种于96孔细胞培养板,每孔100μl细胞悬液(1.0×104个细胞),培养12h后,加入10μl不同浓度的hsb溶液(溶剂为生理盐水),使其终浓度分别为25、50、100、200和400mg/l,对照组则加入相同体积的培养液。每一浓度设4个平行孔,充分混匀,培养48h。每孔加入20μl5mg/mlmtt后继续培养5h。每孔加入100μl三联液(1%sds、5%异丁醇、0.012mol/lhcl,w/v/v),于37℃放置12h后,在570nm波长处测定a值。实验重复3次,计算细胞生长抑制率,细胞生长抑制率=(1–实验组平均a值/对照组平均a值)×100%。测试结果如表4和图13所示。
表4hsb对鸡胚尿囊膜血管生成的抑制率
由表4和图13可知,hsb能够显著抑制人脐静脉血管内皮细胞生长,其抑制效果成剂量依懒性,浓度为400mg/l的hsb对人脐静脉血管内皮细胞生长的抑制率高达90.3%。
应用例2
hsb对cam血管生成的影响:
选择清洁、蛋壳均质、气室均匀的种鸡蛋,用1‰新洁尔灭液擦去污渍,再用75v/v%酒精消毒,随机分为对照组和不同浓度样品处理组,每组10枚,孵育于37.8℃电热培养箱中孵化1周,培养箱中放一水盘以保持相对湿度在40~70%,并留一通气孔,保证氧气的供应。无菌条件下在胚头端开一直径1cm小孔,形成假气室,将不同浓度(0.25、0.5和1mg/ml)的hsb溶液(溶剂为生理盐水)100μl浸过的滤纸置于气室内的尿囊膜上,对照组则加等量pbs,用透明胶带封口。然后置38℃恒温培养箱中培养,24和48h后分别再加hsb溶液100μl于滤纸上,共给药3次。72h后用丙酮和无水乙醇分别固定15min,剪下放有滤纸的膜置载玻片上,弃去滤纸,在显微镜下随机选取5个视野,计算滤纸覆盖范围内可见的血管分支点的数量,以±s表示,血管生成抑制率(%)=(1-给药组血管分支点数/对照组血管分支点数)×100%,计算结果如表5所示。鸡胚尿囊膜血管未经hsb处理的实物图如图14所示,经hsb处理后的实物图如图15所示,其中,a为hsb处理,b为pbs处理。
表5hsb对鸡胚尿囊膜血管生成的抑制率
由表5和图14~15可知,hsb处理的区域的血管细小分支和血管密度明显减少,说明hsb能够显著抑制鸡胚尿囊膜血管生成。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!