氧化亚锡粉末及氧化亚锡粉末的制造方法与流程
本发明涉及一种作为焊锡或电镀等的sn原料使用的氧化亚锡粉末及氧化亚锡粉末的制造方法。
本申请主张基于2015年2月16日于日本申请的专利申请2015-027867号及2016年1月22日于日本申请的专利申请2016-010755号的优先权,并将其内容援用于此。
背景技术:
sn作为在金属材料的表面形成电镀膜的电镀材广泛使用。例如,作为引线框架或连接器等电子组件材料,广泛提供有在由铜或铜合金构成的铜基材的表面实施镀sn或焊锡电镀的电镀铜材料。另外,该电镀铜材料还使用于上述半导体装置中。
并且,在钢板上形成镀sn的马口铁材料一直以来在各种用途中使用。
其中,进行镀sn时,电镀液中的杂质与sn一同析出,由此有可能导致电镀膜的特性发生变化。并且,电镀液中的杂质对镀敷性带来较大影响。因此,要求杂质减少的电镀液。
并且,作为对上述电镀液供给sn的sn供给材料,通常使用氧化亚锡的粉末等。关于该氧化亚锡粉末,要求对于电镀液快速溶解,并且杂质量减少。
因此,专利文献1或专利文献2中,提供有碱量、氯量较少且对于酸为易溶解性的氧化亚锡粉末。
专利文献1:日本特开平11-310415号公报
专利文献2:日本特开2013-079186号公报
然而,关于专利文献1中记载的氧化亚锡粉末,例如如图4所示,呈立方体形状,比表面积比较小,且对电镀液的溶解速度不充分。
并且,关于专利文献2中记载的氧化亚锡粉末,也呈板状或球状的形态,且溶解速度同样不充分。
该发明是鉴于上述内容而完成的,其目的在于提供一种对电镀液等各种酸液的溶解速度快,且作为对电镀液的sn供给材料尤其适合的氧化亚锡粉末及该氧化亚锡粉末的制造方法。
技术实现要素:
为了解决上述课题,作为本发明的一种实施方式的氧化亚锡粉末的特征在于,设为具有朝向外侧突出的多个板状突起部的粒子体,平均粒径设在1μm以上且15μm以下的范围内。
关于如上构成的作为本发明的一种实施方式的氧化亚锡粉末,由于具有朝向外侧突出的多个板状突起部,因此添加到电镀液等时,电镀液等流入板状突起部之间,板状突起部和电镀液等的接触得以促进。并且,该氧化亚锡粉末的平均粒径设在1μm以上且15μm以下的范围内,因此比表面积增大,与电镀液等的接触得以促进。
由此,作为本发明的一种实施方式的氧化亚锡粉末对电镀液等的溶解速度急剧加快,作为对电镀液等的sn供给材料尤其合适。
其中,关于作为本发明的一种实施方式的氧化亚锡粉末,比表面积优选为1.0m2/g以上。
此时,比表面积为1.0m2/g以上而设得比较大,因此与电镀液等的接触得以促进,能够可靠地加快对电镀液等的溶解速度。
并且,关于本发明的一种实施方式的氧化亚锡粉末,体积密度优选在1.5g/cm3以上且小于2.0g/cm3的范围内。
此时,氧化亚锡粉末的体积密度设在上述范围内,因此操作变得容易。
另外,关于作为本发明的一种实施方式的氧化亚锡粉末,优选碱量设为10质量ppm以下,酸量(除碳酸以外)设为50质量ppm以下。
此时,碱量及酸量如上述规定,因此即使作为sn供给材料添加在电镀液等中,也能够抑制电镀液等的组成发生变化。另外,碳酸会发泡而脱出,所以也可以不作为酸量进行评价。
并且,关于作为本发明的一种实施方式的氧化亚锡粉末,优选含有0.2质量%以上的碳酸。
此时,能够与碳酸的发泡一同使氧化亚锡粉末溶解,能够进一步促进对电镀液等的溶解。并且,能够抑制氧化亚锡粉末的氧化。
关于作为本发明的一种实施形态的氧化亚锡粉末的制造方法,其特征在于,其为制造上述氧化亚锡粉末的制造方法,具备:含sn离子酸液形成工序,使酸液中含sn离子而获得含sn离子酸液;第一中和工序,对于所述含sn离子酸液,添加选自碳酸铵、碳酸氢铵、氨水中的任意一种以上的碱液而保持ph3~6,由此获得sn沉淀物;sn沉淀物分离工序,从所述含sn离子酸液分离所述sn沉淀物;sn沉淀物分散工序,使分离的所述sn沉淀物分散在溶剂液中;及第二中和工序,将所述sn沉淀物的分散液维持在50℃以下,并经过1小时以上添加碱液,将ph设为6~12,由此从所述sn沉淀物获得sno。
根据该结构的氧化亚锡粉末的制造方法,能够制造如下的氧化亚锡粉末:设为具有朝向外侧突出的多个板状突起部的粒子体,平均粒径设在1μm以上且15μm以下的范围内,对电镀液等各种酸液的溶解速度快,作为对电镀液的sn供给材料尤其适合。
如上,根据本发明,能够提供一种对电镀液等各种酸液的溶解速度快,作为对电镀液的sn供给材料尤其适合的氧化亚锡粉末及该氧化亚锡粉末的制造方法。
附图说明
图1是作为本发明的实施方式的氧化亚锡粉末(实施例中的本发明例1)的sem观察照片。
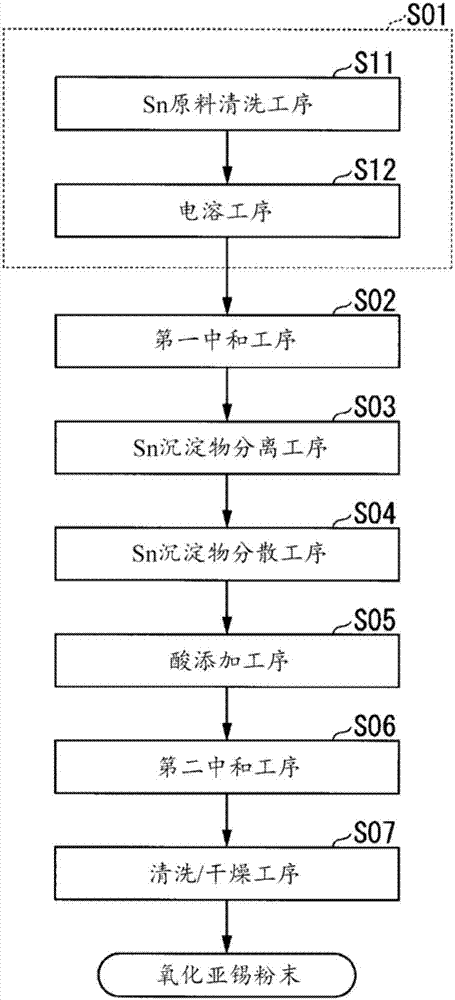
图2是表示图1所示的氧化亚锡粉末的制造方法的流程图。
图3是图2中的第二中和工序的流程图。
图4是以往氧化亚锡粉末的sem观察照片。
图5是实施例中的本发明例7的氧化亚锡粉末的sem观察照片。
具体实施方式
以下,对作为本发明的实施方式的氧化亚锡粉末10及该氧化亚锡粉末10的制造方法进行说明。
本实施方式的氧化亚锡粉末10例如作为对进行镀sn时使用的电镀液的sn供给材料使用。
如图1所示,本实施方式的氧化亚锡粉末10设为具有朝向外侧突出的多个板状突起部11的粒子体,在其外表面,多个板状突起部11相互隔开间隔配置成层状。即,作为本实施方式的氧化亚锡粉末10为配置有朝向外侧突出的多个板状突起部11,且整体呈大致球形的粒子体。另外,优选板状突起部11在一个氧化亚锡粉末10中配置有100片以上。板状突起部11在一个氧化亚锡粉末10中为500片以下为良好,但并不限定于此。
并且,作为本实施方式的氧化亚锡粉末10其平均粒径设在1μm以上且15μm以下的范围内。另外,本实施方式中的平均粒径(d50)设为利用粒度分布测定装置(microtrac公司制造型号名:microtracmt3000粒度分布仪)测定的体积累积中值粒径。
其中,氧化亚锡粉末10的平均粒径小于1μm时,可能导致氧化亚锡粉末10凝聚,并阻碍与电镀液的接触,溶解得不到促进。另一方面,氧化亚锡粉末10的平均粒径超过15μm时,可能导致比表面积不会充分变大,溶解得不到促进而溶解速度变得不充分。
因此,本实施方式中,将氧化亚锡粉末10的粒径设定在1μm以上且15μm以下的范围内。另外,为了抑制氧化亚锡粉末10的凝聚并提高对电镀液的溶解速度,氧化亚锡粉末10的平均粒径的下限优选设为2μm以上。并且,为了增大比表面积并提高对电镀液的溶解速度,氧化亚锡粉末10的平均粒径的上限优选设为10μm以下,进一步优选设为7μm以下。
并且,作为本实施方式的氧化亚锡粉末10中,比表面积设为1.0m2/g以上。氧化亚锡粉末10的比表面积利用bet流动法测定。将氧化亚锡粉末10的比表面积设为1.0m2/g以上,由此能够可靠地提高对电镀液的溶解速度。
为了进一步可靠地提高对电镀液的溶解速度,氧化亚锡粉末10的比表面积优选设为1.5m2/g以上,更优选设为2.0m2/g以上。另外,对氧化亚锡粉末10的比表面积的上限并无特别限定,但优选设为10.0m2/g以下。
并且,体积密度会根据形状的效果而下降,能够设为小于2.0g/cm3,但小于1.5g/cm3时操作性差。
其中,板状突起部11的厚度小于10nm时,或者板状突起部11的厚度超过500nm时,可能导致与电镀液等的接触不充分,且溶解得不到促进。
因此,为了使板状突起部11和电镀液等充分接触,并可靠地促进对电镀液等的溶解,板状突起部11的厚度优选设为10nm以上且500nm以下。另外,板状突起部11的厚度的上限进一步优选设为100nm以下。
其中,本实施方式中,对于板状突起部11的厚度,利用以20000倍的倍率进行sem观察的图像并以标尺条为基准而目测测定并通过计算求出。具体而言,测定sem图像的板状突起部11的厚度,由与标尺条的长度之比计算实际板状突起部11的厚度。
并且,作为本实施方式的氧化亚锡粉末10中,碱量设为10质量ppm以下,酸量(除碳酸以外)设为50质量ppm以下。
本实施方式中,酸量通过制造氧化亚锡粉末10时使用的酸来定义,使用盐酸时成为氯量,使用硫酸时成为硫酸离子量,使用两者时成为其总计值。并且,使用硝酸时的酸量成为硝酸离子量。另外,碳酸会发泡而脱出,所以不作为酸量进行评价。
并且,碱量设为主要残留的氨成分量,na、k为微量,因此作为杂质进行评价。
其中,碱量优选为10质量ppm以下,进一步优选为5质量ppm以下。并且,酸量(除碳酸以外)优选为50质量ppm以下,进一步优选为10质量ppm以下。
另外,在作为本实施方式的氧化亚锡粉末10中,其他杂质也减少,例如na、k、pb、fe、ni、cu、zn、al、mg、ca、cr、mn、co、in及cd的含量分别设为1质量ppm以下。
并且,这些na、k、pb、fe、ni、cu、zn、al、mg、ca、cr、mn、co、in及cd的总计含量设为低于15质量ppm,优选设为低于7.5质量ppm。
其中,pb与sn在特性上近似,是很难从sn分离的元素。并且,na、k是提纯sn原料时有可能混入的元素。fe、ni、cu及zn是从被电镀物或基底电镀层容易混入电镀液中的元素。al、mg、ca、cr、mn、co、in及cd是有可能混入sn原料中的元素。
因此,在作为电镀液的sn供给材料使用的氧化亚锡粉末10中,减少这些杂质元素,由此能够抑制杂质蓄积在电镀液中。
另外,作为本实施方式的氧化亚锡粉末10优选含有0.2质量%以上的碳酸。通过含有0.2质量%以上的碳酸,碳酸发泡的同时氧化亚锡粉末10溶解,由此溶解性进一步提高。并且,通过碳酸,还能够抑制氧化亚锡粉末10的氧化。
其中,为了进一步提高溶解性,并且抑制氧化,优选含有0.3质量%以上的碳酸,进一步优选含有0.5质量%以上。另外,氧化亚锡粉末10中的碳酸的含量的上限并无特别限定,但很难含有超过1.0质量%的碳酸,所以优选设为1.0质量%以下。
接着,参考图2及图3的流程图,对作为本实施方式的氧化亚锡粉末10的制造方法进行说明。
(含sn离子酸液形成工序s01)
首先,使酸液含有sn离子而形成含sn离子酸液。本实施方式中,准备高纯度金属sn(纯度99.99质量%以上),并由酸性洗剂清洗该金属sn的表面(sn原料清洗工序s11)。此时,去除金属sn表面的油分和氧化物,并进行清洗直至金属sn的表面出现金属光泽。
接着,在酸液中电溶已清洗的金属sn,而形成含sn离子酸液(电溶工序s12)。此时,作为酸液,并无特别限定,能够使用甲磺酸、盐酸、硝酸、硫酸、氟硼酸、苯酚磺酸、烷醇磺酸、烷基磺酸等或这些的混合酸。并且,sn的浓度优选例如设在50g/l以上且150g/l以下的范围内,本实施方式中,设为100~110g/l。另外,作为含sn离子酸液也可以使用具有上述酸液的sn电镀液。
(第一中和工序s02)
接着,对于含sn离子酸液添加选自碳酸铵、碳酸氢铵、及氨水中的任意一种以上的碱液而保持ph3~6,由此获得sn沉淀物(氢氧化锡等)。此时,sn作为sn沉淀物(氢氧化锡等)而回收,并且na、k、fe、ni、cu、zn、al、mg、ca、cr、mn、co、in及cd之类的元素残留在含sn离子酸液中。
另外,本实施方式中,将碳酸氢铵水溶液添加至ph成为3.5~4范围。
(sn沉淀物分离工序s03)
接着,从含sn离子酸液分离sn沉淀物(氢氧化锡等)。
(sn沉淀物分散工序s04)
接着,对于已分离的sn沉淀物(氢氧化锡等)反复实施2~3次基于纯水的分散和过滤,来进行sn沉淀物(氢氧化锡等)的清洗。由此,去除附着在sn沉淀物(氢氧化锡等)表面上的杂质。之后,使清洗后的sn沉淀物(氢氧化锡等)分散在纯水中。
(酸添加工序s05)
接着,根据需要,对于使sn沉淀物(氢氧化锡等)分散的分散液添加盐酸或柠檬酸。通过该酸添加工序s05,sn沉淀物(氢氧化锡等)中的第一中和工序s02之前的酸成分被分离。
(第二中和工序s06)
接着,对于使sn沉淀物(氢氧化锡等)分散的分散液,添加碱液并加热,由此从sn沉淀物(氢氧化锡等)获得sno(氧化亚锡)。该第二中和工序s06中,对sn沉淀物(氢氧化锡等)进行脱水,由此形成sno(氧化亚锡)。其中,作为碱液,添加碳酸铵、碳酸氢铵等含有碳酸的物质时,氧化亚锡粉末10中含有碳酸。
本实施方式中,作为碱液添加碳酸氢铵水溶液直至ph成为6以上。
若详细说明,则在该第二中和工序s06中,首先,使sn沉淀物(氢氧化锡等)分散的分散液的温度设为50℃以下(温度调整工序s61)。
对于温度设为50℃以下的分散液,经1小时以上添加碱液,并添加至ph成为6~12范围内(碱添加工序s62)。另外,虽然ph较高也没有问题,但若考虑中和剂的使用量,则ph优选设为6~8左右。并且,若ph过高,则氧化亚锡会溶解,因此设为12以下为较佳。
由此,获得氧化亚锡(sno)。
其中,添加碱液时的分散液的温度超过50℃时,可能导致无法成为具有朝向外侧突出的多个板状突起部11的粒子体。因此,本实施方式中,将添加碱液时的分散液的温度设定在10℃以上且50℃以下。另外,为了可靠地获得具有朝向外侧突出的多个板状突起部11的粒子体,添加碱液时的分散液的温度优选设为30℃以下。并且,以往的方法中,为了促进脱水反应而进行加热,但本发明中,通过两阶段中和,残留酸成分的影响较少,因此脱水反应较快而不需要加热。
并且,碱液的添加时间短于1小时时,可能导致无法成为具有朝向外侧突出的多个板状突起部11的粒子体。因此,本实施方式中,将碱液的添加时间限定在1小时以上。另外,为了可靠地设为具有朝向外侧突出的多个板状突起部11的粒子体,碱液的添加时间优选设为1小时20分钟以上。并且,碱液的添加时间的上限并无特别限定,但从工作效率的观点来看,优选设为2小时以下。
(清洗/干燥工序s07)
接着,对于所获得的sno(氧化亚锡)反复实施2~3次基于纯水的分散和过滤,来进行sno(氧化亚锡)的清洗。由此,去除附着在sno(氧化亚锡)的表面上的铵盐等。之后,过滤/干燥清洗后的sno(氧化亚锡)。
根据以上工序,制造作为本实施方式的氧化亚锡粉末10。
如图1所示,根据作为如上构成的本实施方式的氧化亚锡粉末10,设为具有朝向外侧突出的多个板状突起部11的粒子体,因此添加到电镀液中时,电镀液流入板状突起部11之间,板状突起部11和电镀液的接触得以促进。并且,氧化亚锡粉末10的平均粒径设在1μm以上且15μm以下的范围内,因此比表面积增大。具体而言,能够将氧化亚锡粉末10的比表面积设为1.0m2/g以上。
从以上内容来看,作为本实施方式的氧化亚锡粉末10,能够急剧提高对电镀液等的溶解速度。
另外,本实施方式中,板状突起部11的厚度设在10nm以上且500nm以下的范围内,因此能够可靠地促进板状突起部11和电镀液的接触,从而能够可靠地提高对电镀液的溶解性。
并且,本实施方式的氧化亚锡粉末10中,体积密度设在1.5g/cm3以上且小于2.0g/cm3的范围内,因此操作变得容易。
另外,本实施方式的氧化亚锡粉末10中,碱量设为10质量ppm以下,酸量(除碳酸以外)设为50质量ppm以下,因此即使在电镀液中作为sn供给材料添加,也能够抑制电镀液的组成发生变化。
并且,本实施方式的氧化亚锡粉末10中,例如na、k、pb、fe、ni、cu、zn、al、mg、ca、cr、mn、co、in及cd的含量分别设为1质量ppm以下,因此能够抑制这些杂质元素蓄积在电镀液中,并能够抑制电镀液的劣化。
并且,本实施方式的氧化亚锡粉末10中,含有0.2质量%以上的碳酸,藉此能够与碳酸的发泡一同溶解氧化亚锡粉末10,并能够进一步促进对电镀液等的溶解。并且,通过碳酸的存在而周边的氧气被驱赶,从而成为碳酸气氛,由此能够抑制氧化亚锡粉末10的氧化。
另外,本实施方式中,对于使sn沉淀物(氢氧化锡等)分散的分散液,添加碱液并加热的第二中和工序s06中,添加碱液时的分散液的温度设为50℃以下,碱液的添加时间设为1小时以上,因此能够抑制通过第二中和工序s06生成的氧化亚锡的粒子粗大化,并能够形成具有朝向外侧突出的多个板状突起部11的粒子体。
并且,本实施方式中,具备第一中和工序s02,所述第一中和工序s02中对于含sn离子酸液添加碱液(本实施方式中为碳酸氢铵)而保持ph3~6,由此获得sn沉淀物(氢氧化锡等),因此能够减少sn沉淀物(氢氧化锡等)中的na、k、pb、fe、ni、cu、zn、al、mg、ca、cr、mn、co、in及cd的含量。
而且,由于具备从含sn离子酸液分离该sn沉淀物(氢氧化锡等)的sn沉淀物分离工序s03、使已分离的sn沉淀物(氢氧化锡等)分散在纯水等溶剂液中的沉淀物分散工序s04、及相对于sn沉淀物(氢氧化锡等)的分散液添加碱液并加热,由此从sn沉淀物(氢氧化锡等)获得sno(氧化亚锡)的第二中和工序s06,因此能够有效地获得减少na、k、pb、fe、ni、cu、zn、al、mg、ca、cr、mn、co、in、cd的含量的氧化亚锡粉末10。
并且,本实施方式中,在sn沉淀物分散工序s04和第二中和工序s06之间,具备有对于sn沉淀物(氢氧化锡等)的分散液添加盐酸或柠檬酸的酸添加工序s05,因此即使在sn沉淀物(氢氧化锡等)中含有第一中和工序s02之前的酸成分,也能够去除该酸成分,在之后的第二中和工序s06中能够有效地形成sno(氧化亚锡)。
以上,对本发明的实施方式进行了说明,但本发明并不限定于此,在不脱离其发明的技术思想的范围内能够进行适当变更。
例如,本实施方式中,在含sn离子酸液形成工序s01中,说明了对金属sn进行电溶,但并不限定于此,也可以使用通过其他方法获得的含sn离子酸液。因此,还能够循环利用酸类锡电镀液。
并且,以在sn沉淀物分散工序s04和第二中和工序s06之间具备添加盐酸或柠檬酸的酸添加工序s05进行了说明,但也可以省略该酸添加工序s05。
实施例
以下,对用于确认本发明的有效性而进行的确认实验的结果进行说明。
本发明例1~4及比较例1、2中,作为第一中和工序,在盐酸锡水溶液中添加碳酸氢铵并中和至ph4。第一中和工序中,添加碳酸氢铵时的盐酸锡水溶液的温度、及碳酸氢铵的添加时间设为表1所示的条件。清洗所获得的滤渣而获得sn沉淀物,并用纯水使该sn沉淀物再分散。接着,作为第二中和工序,在sn沉淀物的分散液中添加碳酸氢铵并中和至ph7之后,清洗并干燥所获得的滤渣,由此获得氧化亚锡粉末。另外,第二中和工序中,添加碳酸氢铵时的分散液的温度及碳酸氢铵的添加时间设为表1所示的条件。
在本发明例5中,作为第一中和工序,在硫酸锡水溶液中添加碳酸氢铵并中和至ph4。在第一中和工序中,添加碳酸氢铵时的硫酸锡水溶液的温度及碳酸氢铵的添加时间作为表1所示的条件。清洗所获得的滤渣而获得sn沉淀物,并用纯水使该sn沉淀物再分散。接着,添加盐酸并使sn沉淀物溶解后,作为第二中和工序,添加碳酸氢铵并中和至ph7之后,清洗并干燥所获得的滤渣,由此获得氧化亚锡粉末。另外,在第二中和工序中,添加碳酸氢铵时的分散液的温度及碳酸氢铵的添加时间作为表示1所示的条件。
在本发明例6中,作为第一中和工序,在硝酸锡水溶液中添加碳酸铵并中和至ph4。在第一中和工序中,添加碳酸铵时的硝酸锡水溶液的温度及碳酸铵的添加时间设为表1所示的条件。清洗所获得的滤渣而获得sn沉淀物,并用纯水使该sn沉淀物再分散。接着,添加盐酸并使sn沉淀物溶解后,作为第二中和工序,添加碳酸铵并中和至ph7之后,清洗并干燥所获得的滤渣,由此获得氧化亚锡粉末。另外,在第二中和工序中,添加碳酸铵时的分散液的温度及碳酸铵的添加时间作为表示1所示的条件。
在本发明例7中,作为第一中和工序,在盐酸锡水溶液中添加碳酸氢铵并中和至ph4。在第一中和工序中,添加碳酸氢铵时的盐酸锡水溶液的温度及碳酸氢铵的添加时间作为表1所示的条件。清洗所获得的滤渣而获得sn沉淀物,并用纯水使该sn沉淀物再分散。接着,作为第二中和工序,在sn沉淀物的分散液中添加氨水并中和至ph7之后,清洗并干燥所获得的滤渣,由此获得氧化亚锡粉末。另外,在第二中和工序中,添加氨水时的分散液的温度及氨水的添加时间作为表示1所示的条件。
在本发明例8中,作为第一中和工序,在硫酸锡水溶液中添加碳酸氢铵并中和至ph4。在第一中和工序中,添加碳酸氢铵时的硫酸锡水溶液的温度及碳酸氢铵的添加时间作为表1所示的条件。清洗所获得的滤渣而获得sn沉淀物,并用纯水使该sn沉淀物再分散。接着,作为第二中和工序,在sn沉淀物的分散液中添加碳酸氢铵并中和至ph7之后,清洗并干燥所获得的滤渣,由此获得氧化亚锡粉末。另外,在第二中和工序中,添加碳酸氢铵时的分散液的温度及碳酸氢铵的添加时间作为表示1所示的条件。
在本发明例9中,作为第一中和工序,在硝酸锡水溶液中添加碳酸铵并中和至ph4。在第一中和工序中,添加碳酸铵时的硝酸锡水溶液的温度及碳酸铵的添加时间设为表1所示的条件。清洗所获得的滤渣而获得sn沉淀物,并用纯水使该sn沉淀物再分散。接着,作为第二中和工序,在sn沉淀物的分散液中添加碳酸铵并中和至ph7之后,清洗并干燥所获得的滤渣,由此获得氧化亚锡粉末。另外,在第二中和工序中,添加碳酸铵时的分散液的温度及碳酸铵的添加时间作为表示1所示的条件。
在比较例3、4中,作为中和工序,在硫酸锡水溶液中添加碳酸氢铵并一边加热一边中和至ph7,清洗并干燥所获得的的滤渣,由此获得氧化亚锡粉末。
并且,在比较例5中,作为中和工序,在盐酸锡水溶液中添加碳酸氢铵并一边加热一边中和至ph9,清洗并干燥中和结束后以100℃加热悬浊液并保持1小时而获得的滤渣,由此获得氧化亚锡粉末。
另外,在中和工序中,添加碳酸氢铵时的分散液的温度及碳酸氢铵的添加时间作为表1所示的条件。即,比较例3、4、5中,通过一次中和工序获得氧化亚锡粉末。
对如上述获得的氧化亚锡粉末进行如下评价。
<粒子形状>
以5000倍倍率的sem观察所获得的氧化亚锡粉末,并确认其粒子形状。当所有的粉末成为“具有朝向外侧突出的多个板状突起部的粒子体”时设为“a”,只要有一部分包含板状粒子则设为“b”。将评价结果示于表1。
<氧化亚锡粉末的平均粒径>
将所获得的氧化亚锡粉末的平均粒径(d50)作为利用粒度分布测定装置(microtrac公司制造型号名:microtracmt3000粒度分布仪)测定的体积累积中值粒径进行评价。将评价结果示于表1。
<氧化亚锡粉末的比表面积>
通过bet流动法(macsorbhmmodel-1201)测定所获得的氧化亚锡粉末的比表面积。将测定结果示于表1。
<氧化亚锡粉末的体积密度>
所获得的氧化亚锡粉末的体积密度利用jis体积比重测定器(tsutsuiscientificinstrumentsco.,ltd.制造)并通过定容测定法求出。测定方法的详细说明为,首先,通过称量秤来测量测定容器(不锈钢制容积25ml)的质量。接着,在测定容器中通过筛子(不锈钢制直径2.5mm)加入样品直到溢出。此时,不要对测定容器施加振动,或压缩样品。之后,使用刮平板刮平从测定容器的上端隆起的粉末。此时,刮平板从刮平的方向向后倾斜使用,以便不压缩粉末。最后,通过称量秤测量每个测定容器的质量,扣除测定容器的质量来计算样品的质量,并由测定容器的容积计算出体积密度。将测定结果示于表1。
<氧化亚锡粉末的碳酸量>
通过离子色谱图测定所获得的氧化亚锡粉末中的碳酸量。将测定结果示于表1。
<溶解性的评价>
200ml烧杯(hario公司制造)中加入100g/l的烷基磺酸100ml,在保持成25℃的状态下,投入上述氧化亚锡粉末1g并搅拌,搅拌使用长度30mm的棒状转子(asone公司制造),将搅拌速度设为500rpm。
并且,评价投入的氧化亚锡粉末分散并悬浊化后直至完全溶解而液体变成透明时所需要的时间。将评价结果示于表2。
<碱量及酸量的测定>
通过离子色谱图测定所获得的氧化亚锡粉末中的氨成分并作为碱量。
使用盐酸锡水溶液制造的氧化亚锡粉末中的酸量作为氯量进行评价。氧化亚锡粉末中的氯量通过酸溶解比浊法测定。酸溶解比浊法中,通过分光光度计(hitachi公司制造、u-2910)定量在硝酸水溶液中溶解氧化亚锡粉末,并添加硝酸银水溶液而产生的氯化银的量,求出氧化亚锡粉末中的氯量。
并且,使用硫酸锡水溶液制造的氧化亚锡粉末中的酸量作为硫酸离子量进行评价。使用硝酸锡水溶液制造的氧化亚锡粉末中的酸量作为硝酸离子量进行评价。氧化亚锡粉末中的硫酸离子量和硝酸离子量通过离子色谱图测定。
将测定结果示于表2。
[表1]
[表2]
比较例1中,在第二中和工序中添加碱(碳酸氢铵)时的分散液的温度超过50℃,且所获得的氧化亚锡粉末的一部分未成为具有朝向外侧突出的多个板状突起部的粒子体。因此溶解时间变长,碱量及氯量增加。
比较例2中,第二中和工序中的碱(碳酸氢铵)的添加时间为0.5小时而较短,且所获得的氧化亚锡粉末的一部分未成为具有朝向外侧突出的多个板状突起部的粒子体。因此溶解时间变长,碱量及氯量增加。
比较例3、4中,由一次中和获得氧化亚锡粉末,并未成为具有朝向外侧突出的多个板状突起部的粒子体,例如如图4所示,成为了板状。因此溶解时间变长,碱量及硫酸离子量增加。
比较例5中,由一次中和获得氧化亚锡粉末,并未成为具有朝向外侧突出的多个板状突起部的粒子体,例如如图4所示,成为了板状。因此溶解时间变长,氯量虽减少,但碱量增加。
相对于此,本发明例1~9中,如图1(本发明例1)及图5(本发明例7)所示,可确认到成为具有朝向外侧突出的多个板状突起部的粒子体,且溶解时间较短,溶解性优异。并且,可确认到碱量及酸量(除碳酸以外)较少,且未对电镀液的组成带来较大影响。
并且,本发明例8、9中,未使用盐酸而制造,因此氯量成为检测不到程度的少量。用于制造氯量少的氧化亚锡粉末时,该方法效果最好。
另外,含有0.2质量%以上的碳酸的本发明例1~6及8、9中,与碳酸的含量少于0.05质量%的本发明例7相比,可确认到溶解性进一步提高。
产业上的可利用性
根据本发明,能够提供一种对电镀液等各种酸液的溶解速度快,且作为对电镀液的sn供给材料尤其适合的氧化亚锡粉末及该氧化亚锡粉末的制造方法。
符号说明
10-氧化亚锡粉末,11-板状突起部。
- 还没有人留言评论。精彩留言会获得点赞!