碳包覆铁及碳化铁纳米复合材料及其制备方法与流程
本发明属于碳包覆金属复合材料制备与应用领域,具体涉及一种氮、氧掺杂碳包覆铁及碳化铁纳米复合材料及其制备方法。
背景技术:
金属铁及碳化铁纳米颗粒由于具有优异的光学、电学、磁学性能而备受关注。但金属铁及碳化铁纳米颗粒稳定性差,作为催化剂易发生团聚而失活,此外在液相中容易发生活性相流失,在空气中易被氧化甚至燃烧,极大的限制了此类材料的应用前景。与此同时,碳纳米材料具有耐酸碱腐蚀、化学性质稳定等优点。此外,碳纳米材料在部分反应中还表现出了优异的催化性能。纳米碳包覆金属复合材料是纳米碳与金属复合材料领域的研究热点之一。这类材料由单层至数层弯曲石墨片层为壳紧密包裹金属或金属碳化物纳米颗粒。最新的研究表明,纳米碳材料可以有效保护金属或金属碳化物纳米颗粒,同时由于电子相互作用赋予了复合材料独特的催化性能。因此,这种核壳结构纳米材料在非均相催化、电催化、磁分离、吸波、润滑油添加剂等领域有着广阔的应用前景。
目前,碳包覆金属纳米粒子的方法主要有电弧法、化学气相沉积法(cvd)以及热解法等。其中热解法具有工艺简单、成本低、收率高、金属含量可控等优点,是最具前景的规模化制备碳包覆核壳材料的方法之一。热解法主要可分为两大类,第一类方法直接将氰胺类化合物与无机盐混合后置于惰性或还原气氛下进行高温热解。由于氰胺类化合物易分解,且与金属离子配位作用较弱,导致配体利用率低,用量大,碳化产率低。此外,氰胺类化合物为碳、氮源容易促进副产物碳纳米管的生长。如中国专利cn105478755a公开一种以水溶性金属二价盐、氰胺化合物和含氮、含硼或含硫非金属化合物为前驱体通过裂解法制备碳包覆金属纳米材料的方法。其中金属与氰胺类化合物的摩尔比达到了1:10~20。yao等(waterresearch,2016,101:281-291)以氯化铁、三聚氰胺、硼酸为前驱体经高温热解后得到碳纳米管包覆铁复合材料。另一类方法则是以金属离子与有机配体配位自组装的金属-有机骨架化合物作为前驱体。通常制备这种前驱体需要使用有机溶剂,且需要在反应釜中进行高温、高压反应,步骤繁琐。如yang等(chemicalcommunications,53(27),3882-3885)等以铁氰化钾、聚乙烯吡咯烷酮在盐酸溶液中搅拌制备的普鲁士蓝立方块为前驱体,在ar气氛下高温热解制备了碳包覆碳化铁纳米颗粒。
这两大类热解法具有各自的优点与缺点,若将两类热解法的优点结合在一起,实现纯水相、常压制备前驱体并且减少氰胺配体的用量,对于进一步促进碳包覆金属纳米材料的应用具有重要意义。
技术实现要素:
本发明的目的在于提供一种氮掺杂碳包覆铁及碳化铁纳米复合材料的制备方法。
本发明一方面提供一种碳包覆铁及碳化铁纳米复合材料,包括碳包覆铁及碳化铁纳米颗粒,其中所述碳包覆铁及碳化铁纳米颗粒由铁纳米颗粒和碳化铁纳米颗粒内核和包覆在所述铁纳米颗粒和碳化铁纳米颗粒表面的氮、氧掺杂的石墨化碳层外壳组成;所述复合材料在孔径分布区间2~20nm内具有1个以上的分布峰。
根据本发明的一实施方式,其中以所述复合材料的总质量为基准,fe的含量为15-45%,c的含量为35-80%,n的含量为0.5-12%,o的含量为1-15%,h的含量为0.2-2.5%。
根据本发明的另一实施方式,其中所述碳包覆铁及碳化铁纳米颗粒的粒径为2-35nm。
根据本发明的另一实施方式,其中所述复合材料的酸洗损失率小于40%,优选小于10%。
本发明另一方面提供一种制备碳包覆铁及碳化铁纳米复合材料的方法,包括如下步骤:s1,将柠檬酸配位铁和乙二胺四乙酸配位铁中的一种或多种与尿素、二氰二胺、三聚氰胺和六亚甲基四胺中的一种或多种混合形成前驱体;s2,在惰性保护气氛或还原气氛下高温热解所述前驱体。
根据本发明的一方面,其中所述s1步骤中形成所述前驱体为将fe(oh)3与柠檬酸和乙二胺四乙酸中的一种或多种在水中加热搅拌形成均相溶液,加入二氰二胺、三聚氰胺和六亚甲基四胺中的一种或多种混合形成前驱体溶液,然后除去水分形成所述前驱体。
根据本发明的另一实施方式,其中所述前驱体溶液中fe3+、有机羧酸、氰胺化合物或类似物的摩尔比为1:1~5:1~6。所述有机羧酸为所述前驱体溶液中包含的柠檬酸和乙二胺四乙酸中的一种或多种。所述氰胺化合物或类似物为尿素、二氰二胺、三聚氰胺以及六亚甲基四胺中的一种或多种。
根据本发明的另一实施方式,其中在所述s2步骤中,所述惰性气氛为氮气或氩气,所述高温热解以0.5-30℃/min速率升温至恒温段,在恒温段保持恒温时间为20-600min,所述恒温段温度为400-800℃;优选,所述升温速率为1-10℃/min,在恒温段保持恒温时间为20-600min,所述恒温段温度为500-700℃。
根据本发明的另一实施方式,其中还包括:s3,将所述s2步骤得到的产物在酸性溶液中纯化,除去包覆不完整的铁及碳化铁内核。
根据本发明的另一实施方式,其中所述s3步骤中所述酸性溶液为盐酸、硫酸和氢氟酸的水溶液中的一种或多种,浓度为0.1~3mol/l。
与直接使用无机盐与氰胺类化合物的混合物作为前驱体相比,本发明用有机羧酸配位的铁与氰胺类化合物及类似物混合,有机配体对金属中心具有稳定作为,且本身可作为碳源,因此减少了氰胺类配体及类似物的用量,并且避免了碳纳米管包覆金属副产物的生成。与一般有机配位金属框架化合物前驱体相比,本发明无需使用有机溶剂,利用酸碱反应在水相中配位,金属与配体的配比可调,且常压反应,环境友好,易于规模化制备。本发明制备的氮掺杂碳包覆铁及碳化铁纳米复合材料产品相单一,颗粒分散,碳包覆结构完整,在非均相催化、电催化、磁分离、吸波、润滑油添加剂等领域有着广阔的应用前景。
附图说明
附图是用来提供对本发明的进一步理解,并且构成说明书的一部分,与下面的具体实施方式一起应用于解释本发明,但并不构成对本发明的限制。在附图中:
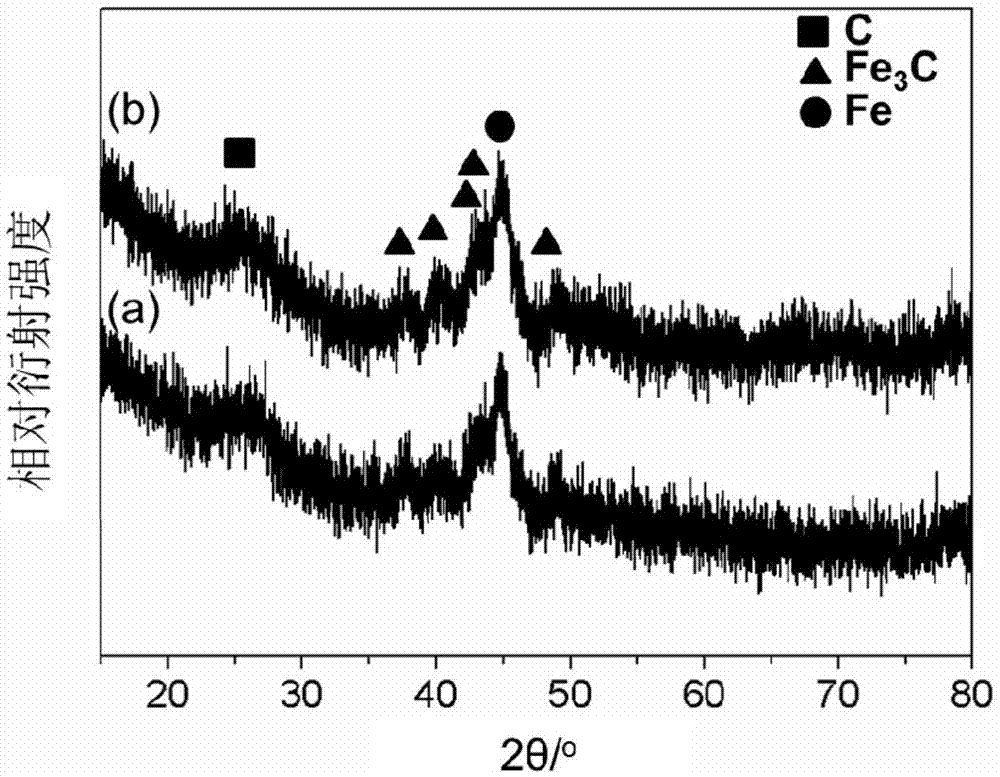
图1是实施例1所制备的氮掺杂碳包覆铁及碳化铁纳米复合材料的xrd图。
图2是实施例1所制备的氮掺杂碳包覆铁及碳化铁纳米复合材料的xps图。
图3是实施例2所制备的氮掺杂碳包覆铁及碳化铁纳米复合材料的tem图。
图4是实施例2所制备的氮掺杂碳包覆铁及碳化铁纳米复合材料的hrtem图。
图5a是实施例2所制备的氮掺杂碳包覆铁及碳化铁纳米复合材料的n2吸脱附等温曲线图。
图5b是实施例2所制备的氮掺杂碳包覆铁及碳化铁纳米复合材料的孔径分布图。
图6是实施例3所制备的氮掺杂碳包覆铁及碳化铁纳米复合材料的xrd图。
图7是实施例3所制备的氮掺杂碳包覆铁及碳化铁纳米复合材料的孔径分布图。。
图8是实施例4所制备的氮掺杂碳包覆铁及碳化铁纳米复合材料的xrd图。
图9是实施例4所制备的氮掺杂碳包覆铁及碳化铁纳米复合材料的孔径分布图。
具体实施方式
以下结合附图通过具体的实施例对本发明作出进一步的详细描述,应当理解的是,此处所描述的具体实施方式仅用于说明和解释本发明,但不以任何方式限制本发明。
本发明中术语“核壳结构”是指内核为铁及碳化铁纳米颗粒,壳层为氮、氧掺杂的石墨化碳层。所述的“石墨化碳层”是指在高分辨透射电镜下可明显观察到“层状的”碳结构,而非无定型结构,且层间距约为0.34nm。
术语“介孔”定义为孔径在2~50nm范围的孔。孔径小于2nm的孔定义为微孔,大于50nm的孔定义为大孔。
术语“氮、氧掺杂”中的“氮”是指氮元素,其中所述纳米复合材料的“氮含量”是指氮元素的含量,具体是指,在碳包覆纳米复合材料制备过程中,形成石墨化碳层中含有以各种形式存在的氮元素,所述“氮含量”为所有形式的氮元素的总含量;同样,“氧”是指氧元素,“氧含量”是指所有形式的氧元素的总含量。
术语“酸洗损失率”是指制备完成的碳包覆过渡金属的纳米复合材料产品经酸洗后过渡金属的损失比例。其反映了石墨化碳层对过渡金属包覆的严密程度。如果石墨化碳层对过渡金属包覆不严密,则在酸处理后,内核的过渡金属会被酸溶解从而流失。酸洗损失率越大,表明石墨化碳层对过渡金属包覆的严密程度越低,酸洗损失率越小,表明石墨化碳层对过渡金属包覆的严密程度越高。
所述的“酸洗损失率”按以下方式测量并计算:
按20ml硫酸水溶液(1mol/l)投加1g样品的比例,在90℃下对样品处理8h,然后用去离子水洗涤至中性,干燥后称重、分析,按下式计算酸洗损失率。
酸洗损失率=[1-(酸洗后复合材料中过渡金属的质量分数×酸洗后复合材料的质量)÷(待酸洗复合材料中过渡金属的质量分数×待酸洗复合材料的质量)]×100%。
本发明的碳包覆铁及碳化铁纳米复合材料,包括碳包覆铁及碳化铁纳米颗粒,其中碳包覆铁及碳化铁纳米颗粒由铁纳米颗粒和碳化铁纳米颗粒内核和包覆在铁纳米颗粒和碳化铁纳米颗粒表面的氮、氧掺杂的石墨化碳层外壳组成;复合材料在孔径分布区间2~20nm内具有1个以上的分布峰。
其中,以复合材料的总质量为基准,fe的含量为15-45%,c的含量为35-80%,n的含量为0.5-12%,o的含量为1-15%,h的含量为0.2-2.5%。
碳包覆铁及碳化铁纳米颗粒的粒径为2-35nm。
复合材料的酸洗损失率小于40%,优选小于10%。
本发明的碳包覆铁及碳化铁纳米复合材料通过如下方法制备,包括如下步骤:s1,将柠檬酸配位铁和乙二胺四乙酸配位铁中的一种或多种与尿素、二氰二胺、三聚氰胺和六亚甲基四胺中的一种或多种混合形成前驱体;s2,在惰性保护气氛或还原气氛下高温热解前驱体。
在s1步骤中形成前驱体的过程,可以为将fe(oh)3与柠檬酸和乙二胺四乙酸中的一种或多种在水中加热搅拌形成均相溶液,加入尿素、二氰二胺、三聚氰胺和六亚甲基四胺中的一种或多种混合形成前驱体溶液,然后除去水分形成前驱体。
优选,前驱体溶液中fe3+、有机羧酸、氰胺化合物或类似物的摩尔比为1:1~5:1~6。其中有机羧酸是指前驱体溶液中含有的柠檬酸和乙二胺四乙酸中的一种或多种。氰胺化合物或类似物是指前驱体溶液中含有的尿素、二氰二胺、三聚氰胺和六亚甲基四胺中的一种或多种。
在s2步骤中,惰性气氛为氮气或氩气,高温热解以0.5-30℃/min速率升温至恒温段,在恒温段保持恒温时间为20-600min,恒温段温度为400-800℃;优选,升温速率为1-10℃/min,在恒温段保持恒温时间为20-600min,恒温段温度为500-700℃。
本发明的制备方法还可以包括:s3,将所述s2步骤得到的产物在酸性溶液中纯化,除去包覆不完整的铁及碳化铁内核。
其中s3步骤中酸性溶液为盐酸或硫酸或氢氟酸水等非氧化性酸的溶液,浓度为0.1~3mol/l。
碳包覆铁及碳化铁纳米复合材料的制备
实施例1
称取10mmolfe(oh)3、15mmol柠檬酸加入100ml去离子水中,在80℃下搅拌得到均相溶液,加入25mmol二氰二胺并继续加热蒸干,将固体研磨后得到前驱体。
将得到的前驱体至于瓷舟内,然后将瓷舟置于管式炉的恒温区,通入氮气,流量100ml/min,并以5℃/min的速率升温至580℃,恒温3h后停止加热,在氮气气氛下冷却至室温,得到碳包覆纳米复合材料。
将得到的复合材料加入60ml1mol/lhcl溶液中,在85℃下搅拌并回流4h后将溶液进行抽滤,并用去离子水洗至中性后将粉末置于100℃烘箱干燥2h,得到纯化的碳包覆纳米复合材料。
实施例2
称取10mmolfe(oh)3、10mmol乙二胺四乙酸加入100ml去离子水中,在80℃下搅拌得到均相溶液,加入30mmol二氰二胺并继续加热蒸干,将固体研磨后得到前驱体。
将得到的前驱体至于瓷舟内,然后将瓷舟置于管式炉的恒温区,通入氮气,流量100ml/min,并以5℃/min的速率升温至600℃,恒温2h后停止加热,在氮气气氛下冷却至室温,得到碳包覆纳米复合材料。
将得到的复合材料加入60ml1mol/lhcl溶液中,在90℃下搅拌并回流4h后将溶液进行抽滤,并用去离子水洗至中性后将粉末置于100℃烘箱干燥2h,得到纯化的碳包覆纳米复合材料。
实施例3
称取10mmolfe(oh)3、25mmol柠檬酸加入100ml去离子水中,在90℃下搅拌得到均相溶液,加入50mmol六亚甲基四胺并继续加热蒸干,将固体研磨后得到前驱体。
将得到的前驱体至于瓷舟内,然后将瓷舟置于管式炉的恒温区,通入氮气,流量100ml/min,并以4℃/min的速率升温至630℃,恒温2h后停止加热,在氮气气氛下冷却至室温,得到碳包覆纳米复合材料。
将得到的碳包覆纳米复合材料加入40ml2mol/lh2so4溶液中,在60℃下搅拌并回流4h后将溶液进行抽滤,并用去离子水洗至中性后将粉末置于100℃烘箱干燥2h,得到纯化的碳包覆纳米复合材料。
实施例4
称取10mmolfe(oh)3、20mmol乙二胺四乙酸加入100ml去离子水中,在70℃下搅拌得到均相溶液,加入50mmol三聚氰胺并继续加热蒸干,将固体研磨后得到前驱体。
将得到的前驱体至于瓷舟内,然后将瓷舟置于管式炉的恒温区,通入氮气,流量100ml/min,并以10℃/min的速率升温至650℃,恒温4h后停止加热,在氮气气氛下冷却至室温,得到碳包覆纳米复合材料。
性能表征
通过xrd获得材料的成分、材料内部原子或分子的结构或形态等信息。所采用xrd衍射仪的型号为xrd-6000型x射线粉末衍射仪(日本岛津),xrd测试条件为:cu靶,kα射线(波长λ=0.154nm),管电压为40kv,管电流为200ma,扫描速度为10°(2θ)/min。
通过高分辨透射电镜(hrtem)表征材料的表面形貌。所采用高分辨透射电镜的型号为jem-2100(日本电子株式会社),高分辨透射电镜测试条件为:加速电压为200kv。样品中纳米颗粒的粒径通过电镜图片测量得到。
通过x射线光电子能谱分析仪(xps)检测材料表面的元素。所采用x射线光电子能谱分析仪为vgscientifc公司生产配备有avantagev5.926软件的escalab220i-xl型射线电子能谱仪,x射线光电子能谱分析测试条件为:激发源为单色化a1kαx射线,功率为330w,分析测试时基础真空为3×10-9mbar。
通过bet测试方法检测材料的孔结构性质。具体采用quantachromeas-6b型分析仪测定,催化剂的比表面积由brunauer-emmett-taller(bet)方法得到,孔分布曲线根据barrett-joyner-halenda(bjh)方法对脱附曲线进行计算得到。
碳(c)、氢(h)、氧(o)、氮(n)四种元素的分析在elementarmicrocube元素分析仪上进行。具体操作方法和条件如下:样品在锡杯中称量1-2mg,放入自动进样盘,通过球阀进入燃烧管燃烧,燃烧温度为1000℃(为了去除进样时大气干扰,采用氦气吹扫),然后用还原铜对燃烧后的气体进行还原,形成氮气、二氧化碳和水。混合气体通过三根解吸柱进行分离,依次进tcd检测器检测。氧元素的分析是利用高温分解,在碳催化剂的作用下,将样品中的氧转化为co,然后采用tcd检测co。
金属元素含量为材料扣除碳、氢、氧、氮含量后归一化结果。
图1是实施例1所制备的氮掺杂碳包覆铁及碳化铁纳米复合材料的xrd图。图中曲线(a)表示未纯化的碳包覆复合材料的xrd谱图;曲线(b)表示纯化后的碳包覆复合材料的xrd谱图。从图中可以看出,纯化前后xrd谱图无明显变化,表明所制备的核壳结构纳米颗粒结构完整,包覆紧密。此外,从xrd谱图中还可以明显看出c、fe3c以及单质fe的衍射峰。图2是实施例1所制备的氮掺杂碳包覆铁及碳化铁纳米复合材料的xps图。可以看出c、n、o、fe四种元素,其原子比为c:n:o:fe=75.72:12.86:10.24:1.18,表明热解过程中大量n元素掺杂进入碳材料结构单元。元素分析仪测定纳米材料c含量为44.25%,h含量为1.68%,n含量为10.07%,o含量为11.61%,归一化后fe含量为32.39%。此外,bet测试表明复合材料孔径分布曲线有两个分布峰,分别位于2.9nm以及3.9nm处。按术语部分所述方法测量、计算,本实施例制得的纯化前的复合材料的酸洗损失率为18%,纯化后的复合材料的酸洗损失率小于1%。在术语部分所述方法的基础上,继续增加酸洗时间,酸洗损失率基本保持不变。
图3是实施例2所制备的氮掺杂碳包覆铁及碳化铁纳米复合材料的tem图。从图中可以看出fe及fe3c纳米颗粒高密度的分散在碳载体上,且颗粒粒径分布集中。图4是实施例2所制备的氮掺杂碳包覆铁及碳化铁纳米复合材料的hrtem图。从图中可以看出fe及fe3c纳米颗粒粒径在10nm左右,并且表层紧密包裹着石墨化碳层。图5a和图5b分别是实施例2所制备的氮掺杂碳包覆铁及碳化铁纳米复合材料的n2吸脱附等温曲线图和孔径分布图。其孔径分布曲线有两个分布峰,分别位于3.0nm以及3.8nm处。元素分析仪测定纳米材料c含量为50.12%,h含量为1.92%,n含量为9.94%,o含量为11.87%,归一化后fe含量为26.15%。按术语部分所述方法测量、计算,本实施例制得的纯化前的复合材料的酸洗损失率为15%,纯化后的复合材料的酸洗损失率小于1%。在术语部分所述方法的基础上,继续增加酸洗时间,酸洗损失率基本保持不变。
图6是实施例3所制备的氮掺杂碳包覆铁及碳化铁纳米复合材料的xrd图。同图1类似,图6中xrd衍射峰分别归属于c、fe3c以及单质fe。图7是实施例3所制备的氮掺杂碳包覆铁及碳化铁纳米复合材料的孔径分布图。其孔径分布曲线有两个分布峰,分别位于3.9nm以及9.5nm处。元素分析仪测定纳米材料c含量为52.90%,h含量为1.69%,n含量为9.46%,o含量为11.14%,归一化后fe含量为24.81%。按术语部分所述方法测量、计算,本实施例制得的纯化前的复合材料的酸洗损失率为24%,纯化后的复合材料的酸洗损失率小于1%。在术语部分所述方法的基础上,继续增加酸洗时间,酸洗损失率基本保持不变。
图8是实施例4所制备的氮掺杂碳包覆铁及碳化铁纳米复合材料的xrd图。同图6类似,图8中的衍射峰同样归属于c、fe3c以及单质fe。图9是实施例4所制备的氮掺杂碳包覆铁及碳化铁纳米复合材料的孔径分布图图。其孔径分布曲线有两个分布峰,分别位于3.9nm以及6.6nm处。元素分析仪测定纳米材料c含量为59.36%,h含量为0.84%,n含量为3.72%,o含量为6.42%,归一化后fe含量为29.66%。按术语部分所述方法测量、计算,本实施例制得的复合材料的酸洗损失率为35%。在术语部分所述方法的基础上,继续增加酸洗时间,酸洗损失率基本保持不变。
与直接使用无机盐与氰胺类化合物的混合物作为前驱体相比,本发明用有机羧酸配位的铁与氰胺类化合物或类似物混合,有机配体对金属中心具有稳定作为,且本身可作为碳源,因此减少了氰胺类配体的用量,并且避免了碳纳米管包覆金属副产物的生成。与一般有机配位金属框架化合物前驱体相比,本发明无需使用有机溶剂,利用酸碱反应在水相中配位,金属与配体的配比可调,且常压反应,环境友好,易于规模化制备。本发明制备的氮掺杂碳包覆铁及碳化铁纳米复合材料产品相单一,颗粒分散,碳包覆结构完整,在非均相催化、电催化、磁分离、吸波、润滑油添加剂等领域有着广阔的应用前景。
当然,本发明还可有其它多种实施例,在不背离本发明精神及其实质的情况下,熟悉本领域的技术人员当可根据本发明作出各种相应的改变和变形,但这些相应的改变和变形都应属于本发明所附的权利要求的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!