一种利用高通量测序构建浮游动物条形码数据库的方法与流程
本发明属于生物技术领域,更具体地说,涉及一种MIT引物设计方法和利用高通量测序构建浮游动物条形码数据库的方法。
背景技术:
DNA条形码(DNA-barcoding)是目前最有力的物种鉴定技术之一,尤其在未知物种的鉴定中起着重要的作用。随着二代测序通量的提高和测序成本的降低,DNA条形码技术开始越来越多的应用在生物多样性调查或环境生物监测中。目前基于DNA条形码的物种鉴定都要以条形码数据库为参照,利用条形码数据库中的物种序列信息来注释待鉴定的序列,所以条形码数据库对DNA条形码而言至关重要。最早的条形码数据库生命条形码协会(Consortium for the Barcode of Life,CBOL)成立于2004年,是最早发起DNA条形码的组织之一,也是目前条形码分类的主要机构,拥有成员200多个,分布于50个国家。数据库中样本数超过八十万,物种数超过七十万种。生命条形码数据库系统(BOLD)是在线的DNA barcode搜集、管理、分析的平台;由管理、分析系(MAS)、识别系统(IDS)和外部连接系统组成(ECS);能对数据进行快速的识别分析。其数据库中目前的barcode序列多达二百七十多万条。NCBI的Genbank也是另一个重要的条形码数据库,包含了数百万条条形码序列。
经检索,中国专利申请公开号CN 102933721 A,申请日为2011年6月8日的专利申请文件公开了用于高通量筛选的组合序列条形码,该发明涉及至少两个核苷酸序列标识符的组合在制备用于高通量测序的样本DNA中的方法和用途,在多种制备的样本DNA的高通量测序中,样本DNA的各制备包括至少两个核苷酸序列标识符的独特组合,其中第一核苷酸序列标识符选自核苷酸序列标识符的一组,且第二核苷酸序列标识符选自核苷酸序列标识符的一组。中国专利申请公开号CN 102877136A,申请日为2012年9月24日的专利申请文件公开了基于基因组简化与二代测序DNA文库构建方法及试剂盒,该方法和试剂盒针对现有文库构建方法不足,可用于参考基因组不完善、研究群体系谱不清晰、无单体型图物种的全基因组SNP检测及基因分型,该发明涉及的DNA文库构建方法及试剂盒操作流程简单,产生的文库测序质量较高。
虽然经过十多年的积累,条形码数据库已经覆盖了大多数物种类群,但是和地球上物种数量相比还是远远不够。现在通用的条形码数据库构建都是基于一代Sanger测序,成本高、准确率低、劳动强度大。对于浮游动物而言,由于其个体较小(50-500μm),纯DNA提取困难,DNA纯度低,往往会导致PCR产量低,由于一代Sanger测序需要的DNA量大(50-500ng),为了满足测序的需要多数情况下需要构建专门的测序载体,通过细菌繁殖增加待测片段的产量,然后进行测序。操作繁琐,而且由于浮游动物是单个体裂解,体内的寄生菌和残留在体内的食物残渣不可避免会带来的DNA污染,PCR产物直接进行一代测序成功率低(往往会出现杂峰),增加条形码数据库的构建的难度。即便正确测序,单个测序反应也只能得到一条序列,效率低下。为了满足日益增长的DNA条形码数据分析的需要,亟待开发一种新的方法能够快速、准确的构建条形码数据库。
技术实现要素:
1.要解决的问题
针对现有技术中条形码构建时操作繁琐、成本高、准确率低以及浮游动物寄生菌和体内食物残渣的干扰等问题,本发明提供一种MIT引物设计方法和利用高通量测序构建浮游动物条形码数据库的方法,相对于Sanger测序,高通量测序技术是一种新型测序技术,可以并行对几十万到几百万条DNA分子进行序列测定,单条序列的测序成本低,可以对一个样本进行深度测序,是潜在进行条形码数据库构建的技术,本发明利用单个体裂解法提取单只浮游动物的DNA,采用特殊的MIT引物进行PCR扩增,PCR扩增产物纯化及定量后可直接进行高通量测序和序列分析,需要的DNA量低,不需要额外的构建测序文库和构建测序载体,操作简单,成本低,后期的序列分析能有效的排除寄生菌和体内食物残渣的干扰,提高了浮游动物条形码数据库构建的准确率。
2.技术方案
为了解决上述问题,本发明所采用的技术方案如下:
一种MIT(线粒体DNA)引物设计方法,其步骤为:
(ⅰ)选取PCR上、下游引物;
(ⅱ)将高通量测序平台的测序接头A连接在PCR上游引物中得到MIT初始上游引物,将高通量测序平台的测序接头P1连接在PCR下游引物中得到MIT初始下游引物;
(ⅲ)设计不同的MIT barcode序列,长度为8-12bp,MIT barcode序列中GC碱基的含量为40-60%;
(ⅳ)将步骤(ⅲ)中设计的MIT barcode插入步骤(ⅱ)中得到的MIT初始上游引物的接头A与PCR上游引物之间得到MIT上游引物。
优选地,所述的步骤(ⅰ)中选取的PCR上、下游引物为细胞色素氧化酶1的上游引物mlCOlinF:5’-GGWACWGGWTGAACWGTWTAYCCYCC-3’和下游引物HCO2198:5’-TAAACTTCAGGGTGACCAAARAAYCA-3’。
优选地,所述的步骤(ⅱ)中高通量测序的平台为Ion torrent测序平台,接头A为5’-CCATCTCATCCCTGCGTGTCTCCGACTCAG-3’,接头P1为5’-CCACTACGCCTCCGCTTTCCTCTCTATGGGCAGTCGGTGAT-3’,所述的步骤(ⅲ)中设计96条不同的MIT barcode序列。之所以选择96条不同的MIT barcode序列是因为:目前市场上的PCR仪多适配96孔板,为便于PCR批量操作,本发明选择设计96条,根据不同实验需要,或者适配特殊的仪器,可随机进行调整。
优选地,所述的步骤(ⅳ)中将步骤(ⅲ)中设计的MIT barcode插入步骤(ⅱ)中得到的MIT初始下游引物的接头P1与PCR下游引物之间得到MIT下游引物。
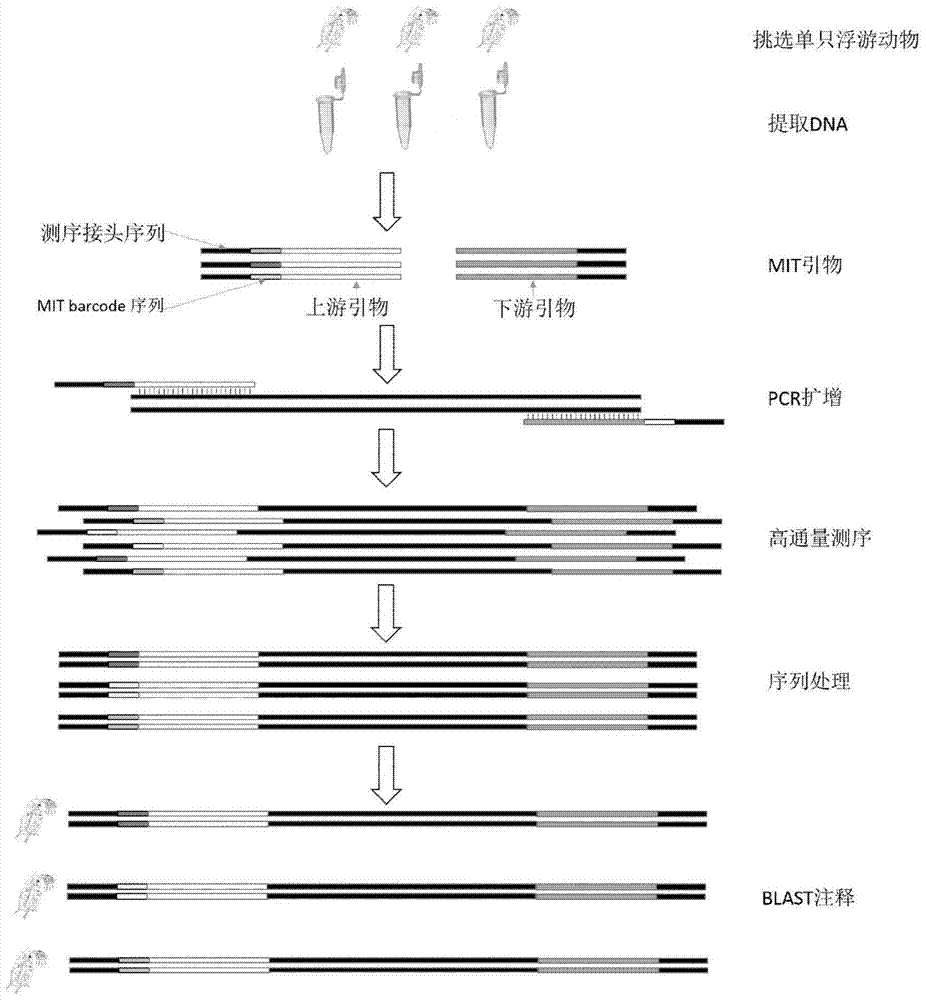
一种利用高通量测序构建浮游动物条形码数据库的方法,其步骤为:
(a)提取DNA:用单个体裂解法提取浮游动物DNA,得到单只浮游动物DNA提取液;
(b)MIT引物PCR扩增:以步骤(a)中得到的单只浮游动物DNA提取液为模板,与上述设计得到的MIT引物进行PCR扩增,反应体系为50μL,由1μL的10μM MIT引物,19μL的去离子水,25μL的2×Mighty Amp buffer,2μL的Mighty Amp DNA聚合酶和2μL的单只浮游动物DNA提取液组成;
(c)PCR扩增产物纯化及定量:用割胶回收试剂盒或者PCR纯化试剂盒对步骤(b)中得到的PCR产物进行回收或纯化,用DNA定量试剂盒对DNA进行定量,纯化、定量后的PCR产物用DNA分析仪进行片段大小分析;
(d)高通量测序:用高通量测序仪对步骤(c)中纯化后的PCR产物进行高通量测序,测序结果以fastq格式输出,过滤测序质量值低和长度短的序列,根据MIT引物中的barcode序列将测序结果分为不同的样本组;
(e)序列分析:根据步骤(d)中得到的不同样本组中的序列分别进行多重序列比对,根据多重序列比对结果,依据遗传距离以0.05为阈值进行序列分组,并去除序列少于5的分组,选取每组中最长的序列作为本组的代表序列并进行BLAST注释;
(f)确定条形码序列:参考物种的分类鉴定信息,选取样品所测序列的注释信息和分类鉴定信息最接近的序列组作为本样品的条形码序列。
优选地,所述的步骤(a)中提取DNA的步骤为:
(1)挑取单只浮游动物,置于EP管中,加入30μL的裂解液,瞬时离心10秒,确保浮游动物浸入裂解液中;
(2)将步骤(1)中瞬时离心后的EP管放置在60-65℃水浴锅中1-2小时,然后瞬时离心10秒,确保管壁上无液体;
(3)将步骤(2)中瞬时离心后的EP管在95℃水浴锅中放置25分钟,然后瞬时离心10秒,确保管壁上无液体;
(4)将步骤(3)中瞬时离心后的EP管在冰上放置3-5分钟,然后瞬时离心10秒,确保管壁上无液体;
(5)向步骤(4)中离心后的EP管中加入30mL的缓冲液,充分涡旋混匀,瞬时离心10秒,确保管壁上无液体,然后将EP管置于-20℃储存。
优选地,所述的步骤(1)中的裂解液为含有25mM的氢氧化钠、0.20mM的乙二胺四乙酸二钠盐、1%的SDS和0.05mg/mL的蛋白酶K的水溶液,PH值为8.0-8.5;所述的步骤(5)中的缓冲液为含有40mM的三羟甲基氨基甲烷盐酸盐的水溶液,PH值为4.5-5.0。
优选地,所述的步骤(e)中通过多重序列比对计算序列间的遗传距离,并将序列根据遗传距离从小到大排序,以0.05为阈值进行序列分组,剔除序列数小于5的分组。
优选地,所述的步骤(e)中当BLAST注释序列相似性大于或等于97%时,将BLAST返回序列作为注释序列进行注释。
优选地,所述的步骤(e)中当BLAST注释序列相似性小于97%时,选取BLAST返回的前50条序列依据序列间的遗传距离构建NJ系统进化树,以进化树中最近似的序列作为注释序列。
3.有益效果
相比于现有技术,本发明的有益效果为:
(1)本发明中采用单个体裂解法提取浮游动物DNA,能有效的提取浮游动物的DNA,特有的SDS的蛋白酶K可以提高DNA纯度,最大程度的减少蛋白质对PCR反应的抑制作用,很好的保证了后续PCR的顺利进行;
(2)本发明中采用专门的MIT引物(包含测序接头序列、8-12个碱基的barcode序列和常规PCR引物),其中的测序接头序列使得PCR产物经过纯化定量后可直接进行测序,减少了构建测序文库的步骤,降低测序成本的同时也减少了操作时间,其中的8-12个碱基的barcode序列能够将不同的样本区分开来,使得一次测序可以测多个样本,提高了测序效率;
(3)本发明中采用高通量测序技术来构建条形码数据库,一次可获得大量条形码序列,即便其中有浮游动物寄生菌和体内食物残渣的干扰,也能通过后续的序列分析除去,增加了条形码数据库构建的成功率;
(4)本发明开发了专门的序列分析方法,原理为:根据序列间的遗传距离从小到大排序,以0.05为阈值进行序列分组,能够有效的区分目的序列和干扰序列(寄生菌和食物残渣),增加了数据的准确性,基于以上原理的运算可在各种不同的平台中实现,操作简便,运算速度快,便于进行自动化处理,减少了人工筛选的时间,提高了计算效率;
(5)本发明利用本地BLAST程序对测序数据进行注释,当BLAST返回的注释序列相似性大于等于97%时,可直接注释到物种水平,当BLAST注释序列相似性小于97%时,挑选BLAST返回的前50条序列依据序列间的遗传距离构建NJ系统进化树,以进化树中最近似的序列作为注释序列,增加了序列注释的准确性和提高序列的利用率。
附图说明
图1为本发明利用高通量测序构建浮游动物条形码数据库的示意图;
图2为本发明中MIT引物构成示意图;
图3为本发明中部分样品MIT引物PCR扩增图;
图4为本发明中Sample 20样本中所有序列按照遗传距离排序图;
图5为本发明中依据序列遗传距离构建的NJ系统进化树,标*的为待注释序列。
具体实施方式
在本发明中所使用的术语,除另有说明外,均为本领域普通技术人员通常理解的含义。
下面结合具体实施例,并参照数据进一步详细地描述本发明。应理解,这些实施例只是为了举例说明本发明,而非以任何方式限制本发明的范围。
在以下的实施例中,未详细描述的各种过程和方法是本领域中公知的常规方法。所用试剂的来源、商品名以及有必要列出其组成成分者,均在首次出现时标明,其后所用相同试剂如无特殊说明,均以首次标明的内容相同。
实施例中所述的引物都由上海捷瑞生物工程有限公司合成,使用前溶解于去离子水中,浮游动物为2014年8月份采集于江苏太湖浦庄、大浦口、西山和庙港(采集过程进行了保密,未公开)等位点,PCR试剂为Mighty Amp DNA Polymerase(Takara),PCR仪器为bio-rad热循环仪。
下面结合具体实施例对本发明进一步进行描述。
实施例1
本发明适用于各种高通量测序平台,本实例仅以Ion Torrent测序平台为例设计MIT引物,其他测序平台只需更换各自的测序接头序列即可。细胞色素氧化酶1(CO1)为动物中最通用的条形码标签,也是条形码数据库中重要的组成序列。因此本实例就以CO1为例设计MIT引物。通用的CO1上、下游引物分别为mlCOlinF(5’-GGWACWGGWTGAACWGTWTAYCCYCC-3’)和HCO2198(5’-TAAACTTCAGGGTGACCAAARAAYCA-3’)。IonTorrent高通量测序平台的测序接头序列分别为A adapter(5’-CCATCTCATCCCTGCGTGTCTCCGACTCAG-3’)和P1adapter(5’-CCACTACGCCTCCGCTTTCCTCTCTATGGGCAGTCGGTGAT-3’),将A adapter连接在上游引物中,P1adapter连接在下游引物上。综上,用于Ion Torrent高通量测序平台的MIT引物如图2所示。我们设计了96种不同的MIT barcode,长度为8-12bp,其碱基组成如表1所示。在A adapter和mlCOlinF之间插入MIT barcode形成96条MIT-CO1-上游引物。P1adapter连接HCO2198形成下游引物(根据不同研究的需要,也可以在下游引物也插入MIT barcode序列,形成不同的下游引物)。在进行PCR反应时,每一个样本选用不同的上游引物,后期的测序结果可以根据上游引物上的barcode序列辨别本序列来自哪一个样本。
表1 MIT barcode序列的碱基组成
如图1所示,在倒置显微镜下随机挑选一只浮游动物,根据相应的形态学特征进行物种鉴定,必要时进行解剖。鉴定完毕后,对物种进行拍照备份,然后在体视镜下小心将浮游动物转移到0.2mL的EP管中,加入30μL的裂解液(含有25mM的氢氧化钠、0.20mM的乙二胺四乙酸二钠盐、1%的SDS和0.05mg/mL的蛋白酶K的水溶液,PH值为8.0-8.5),瞬时离心10秒,确保浮游动物浸入裂解液中;将EP管在60-65℃水浴锅中放置1小时,瞬时离心10秒,确保管壁上无液体;然后转移至95℃水浴锅中放置25分钟,瞬时离心10秒,确保管壁上无液体;结束后即刻将离心管在冰上放置4分钟,瞬时离心10秒,确保管壁上无液体;最后加入30μL的缓冲液(含有40mM的三羟甲基氨基甲烷盐酸盐的水溶液,PH值为4.5-5.0),充分涡旋混匀,瞬时离心10秒,确保管壁上无液体,-20℃储存备用。
采用Takara公司的Mighty Amp DNA聚合酶进行PCR扩增。反应体系为50μL,包含以下组分:1μL的10μM的实施例1中上下游MIT-CO1-引物,19μL的去离子水,25μL的2×Mighty Amp buffer,2μL的Mighty Amp DNA Polymeras,2μL的单只浮游动物DNA提取液。PCR反应条件如表2所示。PCR产物用浓度为1.5%的琼脂糖胶检测。图3为部分样品的琼脂糖胶检测图。结果表明:PCR扩增条带单一明亮,无非特异性扩增,无明显拖尾,表明MIT引物可以很好的进行PCR扩增,插入的MIT barcode序列和测序接头不会对PCR扩增效果造成太大的影响,所选的1520个样本中,成功扩增出1323个,扩增成功率为87%,完全满足后续测序的要求。
表2 Touchdown PCR反应程序
实施例3
实施例2中的PCR扩增产物用MinElute Gel Extraction Kit(Qiagen,USA)试剂盒进行割胶回收,用QubitTM dsDNA HS Assay Kits进行定量,纯化后的PCR产物用Bioanalyser 2100(Agilent Technologies,USA)进行片段大小分析。采用Ion Torrent PGM测序仪进行高通量测序。测序结果以fastq格式输出,在基于Ubuntu系统的QIIME平台下进行序列的前处理,过滤测序质量值低和长度短的序列,根据MIT barcode序列将测序结果分为不同的样本组。在测序的1323个样本中成功测出1257个,检出率高达95%。以Sample 20为例,在R语言“DECIPHER”软件包中对Sample 20所有序列进行多重序列比对,序列间的遗传距离从小到大排序如图4所示,可以看出序列分为3个大的类群,以0.05为阈值将序列分成3组,每一组中随机抽取一个序列作为本组的代表序列。序列明显分成3个类群说明存在污染序列,这在浮游动物中比较常见,在本次测序的所有的样本中,出现污染序列的比例为38%。
实施例4
利用本地BLAST程序对实施例3中的代表序列进行注释,当BLAST返回的结果中序列相似性大于97%时,则直接以返回序列进行注释。当BLAST返回的结果小于97%时,选取BLAST返回的前50个序列和待注释序列进行多重序列比对,依据序列间的遗传距离构建NJ系统进化树,如图5所示,选取在进化树上与带注释序列距离最近的序列作为注释序列。根据此方法,实施例3中Sample 20的3个类群分别来自细巧华哲水蚤、盔形溞和泥鳅。而Sample20本身经过鉴定是汤匙华哲水蚤,据此判断Sample 20的条形码序列应该是第一类群。两外两个类群均属于干扰序列。通过上述序列分析方法,可有效的区分污染序列和组成序列,增加了条形码数据库的准确性。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种构建Hi‑C高通量测序文库的方法与制造工艺
- 桑葚病原菌高通量鉴定及种属分类方法及其应用与制造工艺
- 基于高通量测序技术检测乳腺癌/卵巢癌易感基因的多重PCR特异性引物、试剂盒及方法与制造工艺
- 基于高通量测序检测人EGFR基因变异的质控方法及试剂盒与制造工艺
- 基于高通量测序技术的小麦病原微生物检测方法及其应用与制造工艺
- 一种基于二代高通量测序技术的地中海贫血症的基因检测试剂盒的制造方法与工艺
- 一种基于集群的高通量数据分析方法与制造工艺
- 高通量双折射干涉成像光谱仪装置及其成像方法与制造工艺
- 高通量测序检测人循环肿瘤DNA EGFR基因的捕获探针及试剂盒的制造方法与工艺
- 一种高通量测序检测无创亲子鉴定的方法与制造工艺