一种基于集群的高通量数据分析方法与流程
本发明属于基因测序技术领域,特别涉及一种基于集群的高通量数据分析方法。
背景技术:
高通量基因测序技术又称“下一代”测序(next-generation sequencing,NGS)技术,可以一次性测定几十万甚至几百万条序列,是现今应用最广泛的测序技术。相对于传统的Sanger测序技术,NGS具有高速、高通量、低价格等优点。
基于高通量测序的变异检测在近年发展迅猛,现今又恰逢精准医疗的大力推广,变异检测的需求面临爆发式的增长。在变异检测的过程中,处理GB级别甚至上百GG的数据量是很普遍的情形,常规分析时间从几个小时到几天不等,医生或病人获知检测报告的时间较长,是精准医疗面临的一个问题。
除了提高硬件性能外,常用的加速变异检测的方式是使用多线程的方式对数据进行处理,然而基于应用程序多线程的加速处理对分析速度的提升有限,并且有所限制,如果应用程序不支持多线程,则此种加速则无法进行。
现今有基于分布式文件系统的Map Reduce加速方法,效果是很好的,但是这种方法需要对分布式文献系统有所了解,需要掌握相应的对口语言编程(比如java),部署较为困难,使用相较复杂,不利于这种并行计算方式在分析高通量测序数据方面的实施。
申请号CN201510192260.8的专利文献,涉及“一种超快速检测人类基因组单碱基突变和微插入缺失的方法,是一种能从人基因组DNA测序结果中快速地检测出单碱基突变、微插入缺失的可行方法”。该申请“通过把人参考基因组序列科学有效地切分为小的子参考序列块,把人重测序中的几乎全部步骤(包括分析时间较长的步骤)都切分为计算复杂度大大降低的子任务块,而各子任务块之间相互不影响,最后把从各子参考序列块中得到的遗传多态性信息进行去冗余、校正,然后过滤,从而得到原人重测序流程中需要获取的遗传多态性信息”。该申请认为上述方案解决了人重测序生物信息分析时间过长的问题,然而,该文献对于高通量基因测序技术并没有涉及,没有给出解决现有高通量基因测序方案中所存在问题的建议。
可见,现有的方案只能作全基因组数据的并行处理,对于全外显子或目标测序的情形不能较好应对,另外如果并行处理的任务数做了改变,需要重新分割参考基因组并作索引,增加了分析的时间和不便。
技术实现要素:
本发明提供一种基于集群的高通量数据分析方法,该方法也是一种用于变异检测的通用简易的并行计算方法,用于加速整个分析过程。
本发明的技术方案是,一种高通量数据分析方法,对高通量测序下机数据的处理包括:
对下机数据进行数据分割;
对于分割后获得的多个数据片的运算处理,采用集群管理工具分配包括计算节点,以及相应的CPU和内存的计算资源。
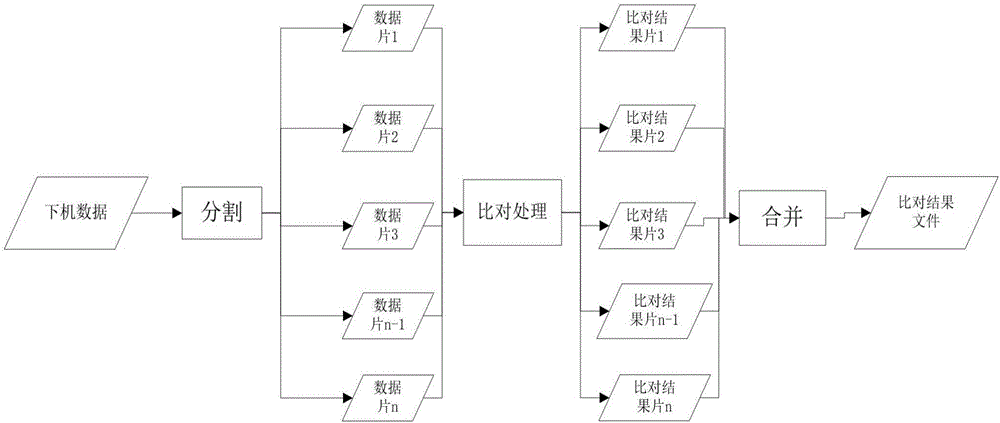
优选的,在对下机数据进行数据分割后,生成多个前数据片文件,在与参考基因组的所有比对完成后,将生成的多个比对结果片文件合并为一个比对结果文件;
预先指定一个区域文件,将其分割成指定的多个区域子文件;
将所述比对结果文件根据指定的多个区域子文件抽取数据进行再次分割,生成多个后数据片文件,提供给后续步骤处理。
前数据片文件和后数据片文件均为以行为每条记录单位的区隔,做分割处理时,预先设定文件的总行数,以此控制产生的片文件数量,也由此设定需要并行处理的任务数。
优选的,该分析方法用于变异检测,测序下机数据文件为fastq格式。
优选的,该分析方法运行于linux系统,使用linux shell编程,集群管理工具使用torque。
一种高通量数据分析方法,包括以下步骤:
(1)对高通量测序下机数据进行数据分割,测序原始数据为fastq格式,下机数据是经过压缩的;
(2)使用torque对分割的数据分配计算节点、CPU和内存,作剪切adaptor序列、末端无效序列、低质量末端序列的处理,结果数据格式为fastq;
(3)使用torque对步骤(2)获得的结果数据分别分配计算节点、CPU和内存,将reads比对到参考基因组,结果数据格式为sam;
(4)使用torque对步骤(3)获得的结果数据分别分配计算节点、CPU和内存,对比对结果文件作的处理包括,对比对结果进行排序、去除比对质量低的部分比对结果、将比对结果数据作压缩和对比对结果作索引,结果数据格式为bam;
(5)使用torque对步骤(4)获得的结果数据分配计算节点、CPU和内存,将所有比对结果文件整合为一个总比对文件,结果数据格式为bam;
(6)使用预先指定的目标区域文件,将此目标区域文件包含的区域分割成指定的多个目标区域子文件,目标区域文件使用bed格式;
(7)按照各自的目标区域文件,使用torque对将步骤(5)获得的结果文件分配计算节点、CPU和内存,分割比对文件重新分割为多个文件,每个文件的比对数据比对到的区域只包含各自目标区域,结果数据格式为bam;
(8)使用torque对步骤(7)获得的结果数据分别分配计算节点、CPU和内存,作去除PCR引起的重复序列的处理,结果数据格式为bam;
(9)使用torque对步骤(8)获得的结果数据分别分配计算节点、CPU和内存,作indel区域再比对的处理,结果数据格式为bam;
(10)使用torque对步骤(9)获得的结果数据分别分配计算节点、CPU和内存,作碱基质量值再校正的处理,结果数据格式为bam;
(11)使用torque对步骤(10)获得的结果数据分别分配计算节点、CPU和内存,作SNP calling和INDEL calling,结果数据格式为vcf;
(12)使用torque对步骤(11)获得的结果数据分别分配计算节点、CPU和内存,对变异进行过滤和注释,结果数据格式为vcf;
(13)使用torque对步骤(12)获得的结果数据分别分配计算节点、CPU和内存,将所有的变异结果文件进行整合,结果数据格式为txt、xls、pdf或html。
本发明通过提供一个区域文件来定位各个任务对应的参考基因组的区域,能应对全基因组,全外显子,目标测序等,并很方便地扩展到其他分析上,比如拷贝数变异的分析。同时本发明还能灵活指定分析的并行处理任务数,根据任务数将提供的目标区域作分割,定位并行处理各自对应的参考基因组的区域,这可以灵活充分利用计算机资源。
用本发明的方法对高通量基因测序数据进行数据分割和并行化处理和分析,极大提高了分析速度。比如说,如果将原始数据分割成十份数据同时处理,在良好的情形,可以减小分析时间接近于原来的十分之一,因为分析时会有数据分割和再整合的过程。计算资源越多,分析数据越大,可以将数据分割为更多部分并行处理,本方法的效果更为明显。
附图说明
图1是本发明的数据分析方法中部分流程示意图。
图2是本发明的数据分析方法中部分流程示意图。
具体实施方式
如图1所示,对高通量测序下机数据进行数据分割。在对下机数据进行数据分割后,产生n个数据片文件。这些数据片文件在与参考基因组的所有比对完成后,将生成的n个比对结果片文件合并为一个比对结果文件。
如图2所示,预先指定一个区域文件,将其分割成指定的n个区域子文件。将所述比对结果文件根据指定的n个区域子文件抽取数据进行再次分割,生成n个数据片文件,提供给后续步骤处理。
上述的数据片文件均为以行为每条记录单位的区隔,做分割处理时,预先设定文件的总行数,以此控制产生的片文件数量,也由此设定需要并行处理的任务数。
对于分割后获得的多个数据片的运算处理,采用集群管理工具分配包括计算节点,以及相应的CPU和内存的计算资源。
本发明提供的一个实施例如下。
一种基于集群的高通量数据分析加速方法,用于变异检测,其步骤包括,(优选的,运行平台为linux,优选的,主程序使用linux shell编程。计算机集群系统基于hadoop框架。优选的,集群管理系统使用torque):
1)对高通量测序下机数据进行数据分割;优选的,所述测序原始数据为fastq格式,下机数据通常经过压缩。比如经由gzip压缩的数据后缀通常写作.fastq.gz。优选的,数据分割使用linux系统下的命令cat,zcat,gzip,pigz,split实现。
2)使用集群管理工具对分割的数据分别分配计算节点,CPU,内存作剪切adaptor序列,末端无效序列,低质量末端序列的处理,同时监测各个小任务的完成情况;优选的,中间结果数据格式为fastq,使用工具cutadapt。
3)待2)中相应小任务处理完成,使用集群管理工具对2)的中间结果数据分别分配计算节点,CPU,内存,将reads比对到参考基因组,同时监测各个小任务完成情况;优选的,中间结果数据格式为sam,使用工具bwa mem。
4)待3)中相应小任务处理完成,使用集群管理工具对3)的中间结果数据分别分配计算节点,CPU,内存,对比对结果文件作相关处理:对比对结果进行排序,去除比对质量低的部分比对结果,将比对结果数据作压缩,对比对结果作索引。同时监测各个小任务完成情况;优选的,中间结果数据格式为bam,使用工具samtools view,samtools sort,samtools index。
5)待4)中各个任务处理完成,使用集群管理工具对4)的所有中间结果数据分配计算节点,CPU,内存,将所有比对结果文件整合为一个文件。同时监测此任务完成情况;优选的,中间结果数据格式为bam,使用工具samtools merge,samtools index。
6)使用预先指定的目标区域文件,将此目标区域文件包含的区域分割成指定的多个目标区域文件。同时监测各个小任务完成情况;优选的,目标区域文件使用bed格式,分割目标区域使用python语言编写程序实现。
7)待6)中任务处理完成,按照各自的目标区域文件,将5)中的结果文件分配计算节点,CPU,内存,重新分割为多个文件,每个文件的比对数据比对到的区域只包含各自目标区域。同时监测各个小任务完成情况;优选的,中间结果数据格式为bam,使用samtools view-L分割比对文件。
8)待7)中任务处理完成,使用集群管理工具对7)的中间结果数据分别分配计算节点,CPU,内存,分别作去除PCR等引起的重复序列的处理。同时监测各个小任务完成情况;优选的,中间结果数据格式为bam,使用picard MarkDuplicates.jar工具作处理。
9)待8)中任务处理完成,使用集群管理工具对8)的中间结果数据分别分配计算节点,CPU,内存,分别作indel区域再比对的处理。同时监测各个小任务完成情况;优选的,中间结果数据格式为bam,使用gatk RealignerTargetCreator,gatk IndelRealigner工具作处理。
10)待9)中任务处理完成,使用集群管理工具对9)的中间结果数据分别分配计算节点,CPU,内存,分别作碱基质量值再校正的处理。同时监测各个小任务完成情况;优选的,中间结果数据格式为bam,使用gatk BaseRecalibrator,gatkPrintReads工具作处理。
11)待10)中任务处理完成,使用集群管理工具对10)的中间结果数据分别分配计算节点,CPU,内存,分别作SNP calling和INDEL calling。同时监测各个小任务完成情况;优选的,中间结果数据格式为vcf,使用gatk UnifiedGenotyper工具作处理。
12)待11)中任务处理完成,使用集群管理工具对11)的中间结果数据分别分配计算节点,CPU,内存,对变异进行过滤和注释。同时监测各个小任务完成情况;优选的,中间结果数据格式为vcf,使用gatk VariantFiltration,gatk CombineVariants,snpEff,snpsift,VEP,annovar工具作处理。
13)待12)中任务处理完成,使用集群管理工具对12)的中间结果数据分别分配计算节点,CPU,内存,将所有的变异结果文件进行整合。优选的,结果数据格式为txt,xls,pdf,html等。使用perl,python,R,matlab等语言编写程序进行处理。
根据上述实施例,以全外显子分析为例,通过指定每个文件的总行数的数值,我们能控制分割的文件的个数。如果计算机资源较多,我们可以指定较小的总行数数值,生成较多的文件数作并行处理,反之可以指定较大的总行数数值。
以此我们首先将原始fastq文件分割成多个fastq文件,再将分割后的文件通过并行处理的方式分别比对到参考基因组,接下来我们将比对的文件合并成一个文件并排序。之后,我们将全外显子区域文件分割成指定个数的多个区域文件,每个区域文件包括全外显子区域的部分区域。通过分割成的各个区域文件,我们从合并成的比对文件抽取对应区域的比对数据,形成多个比对文件。接下来,我们对这些比对文件并行处理,加快分析速度。如果计算机资源较多,我们可以指定较大的并行处理任务数目,如果计算机资源较少,我们指定少的任务数。另外,依次进行的各个分析通常使用不同的计算机资源,通过集群管理系统的按需调配,提高了资源利用率。
另外,流程中大量使用了gatk集中的多个多个工具,通过人为限定分析时所对应的区域,能另外加快程序运行,在理论中更能提高分析的准确度。以全基因组的分析时间来看,从比对到基因型检测,再到注释和过滤,我们有记录的结果是把大于一周的分析时间,缩短到十二个小时完成,速度是高于原来的十四倍。
- 还没有人留言评论。精彩留言会获得点赞!