核酸酶介导的编辑编码HLA的基因所产生的免疫相容性细胞的制作方法
本发明涉及一种用于产生免疫相容性细胞或细胞群的方法,所述方法包括:在分离的细胞中,通过基因缺失或基因修饰来编辑一种或多种免疫相容性抗原基因的一个或两个等位基因,所述分离的细胞包括选自HLA(人类白细胞抗原)-A、HLA-B和HLA-DR的至少一种免疫相容性抗原基因;本发明还涉及通过上述方法产生的免疫相容性细胞,以及包括通过上述方法产生的免疫相容性细胞的细胞群。
背景技术:
人类胚胎干细胞(hESC)是能够无限自我更新的多能细胞,并且可以分化成所有三个胚层的细胞类型。由于在适当的条件下,hESC响应于外部信号并且能够被诱导而分化成特定的细胞类型(诸如功能性心肌细胞和胰腺β细胞),所以hESC的首次使用预示着再生医学迈入新时代。这些特性使得人类胚胎干细胞成为再生医学中宝贵的细胞资源。患有神经退行性疾病、自身免疫疾病、心血管疾病和造血疾病的患者是干细胞疗法的潜在受益者。尽管人类胚胎干细胞具有巨大的潜势,但临床中异源hESC衍生细胞的免疫排斥是该细胞应用的主要障碍。人类白细胞抗原(HLA)的细胞表面表达是主要的免疫屏障,其中,人类白细胞抗原在主要组织相容性复合体中通过基因编码。
通过进行体细胞核转移(SNT)或者使能够诱导体细胞成为被称为诱导多能干细胞(iPSC)的ESC样细胞的4种转录因子Oct4(O)、Sox2(S)、Klf4(K)和c-Myc(M)强制表达可在体细胞中重建hESC的独特性质。这两种方法能够使患者衍生出特异性多能干细胞。重要的是,由于这些细胞得自于患者自身的细胞,所以认为他们的免疫系统不会排斥这些细胞。
然而,由于SNT介导的人类干细胞的重编程处于初期阶段、具有较低的效率并且需要卵母细胞移植技术,所以还不能提供实用的解决方案。尽管根据目前的报道,似乎有望相对简单地衍生出iPSC,但是基于转录因子的重编程与不完全的表观重编程相关。因此,在临床中使用这些细胞需要详细检查iPSC克隆体,这是繁琐的。此外,在良好作业规范(GMP)下,对于个体患者来说,产生多能干细胞在经济上上很可能行不通。
克服干细胞移植的免疫屏障的一种方法是建立具有HLA单倍型的临床级hESC/iPSC/SNT-hESC库,其将匹配于相当大比例的人群。然而,在当前良好作业规范(cGMP)下,hESC/iPSC/SNT-hESC系的衍生需要投入大量金钱和时间。
避免hESC衍生物的免疫排斥的另一方案是HLA分子的基因操作。Torikai和同事利用锌指核酸酶,在ESC中选择性地消除了人类白细胞抗原(HLA)I类,并且证实了HLA-A细胞能够免受HLA-限制性细胞毒性T淋巴细胞的裂解。然而,HLA I类完全敲除的细胞为NK细胞介导的细胞毒性的靶标(Oberg L etc.,Eur J Immunol.,2004,34(6):1646-1653)。
在另一项研究中,Riolobos和同事对β-2微球蛋白(B2M)进行阻断,其编码主要组织相容性复合体(MHC)I类分子的附属链,并且需要他们来用于表面表达(Laura Riolobos etc.,2013,21(6):1232-1241)。因此,B2M基因的纯合缺失防止I类HLA分子的表面易位,并且降低免疫原性。然而,据报道,缺少B2M基因的造血干细胞被NK细胞消除,所以该方法提供了的方案是受限的。尽管,在体外,较低多态性的HLA-E或HLA-G-B2M嵌合蛋白的强制表达保护了I类阴性细胞免受NK细胞介导的裂解,但是还没有关于体内数据的任何报道。
最近,已经表明在人源化小鼠中,细胞毒性T淋巴细胞相关的蛋白4(CTLA4)免疫球蛋白和程序性细胞死亡配体1(PDL1)敲入的人ESC(hESC)同时阻断T细胞共刺激途径并且激活T细胞抑制途径。众所周知的是,感染的细胞同样进行免疫应答。没有体内研究表明在CTLA4和PDL1双重敲入细胞被感染时,免疫细胞能否将这些细胞消除。
看起来HLA分子的单等位突变和HLA纯合的hESC的后续衍生可能提供了用于解决hESC的免疫排斥的方案。由现存hESC系得到的纯合hESC的小文库可覆盖相当大百分比的人群。
技术实现要素:
技术问题
本发明人进行深入的研究来开发携带半合型HLA基因的用于移植的细胞,从而不会在受体中引起免疫排斥;以及一种用于构建包括该细胞的细胞群的方法。因此,本发明人发现,通过对HLA-1基因、HLA-B基因和HLA-DR基因,也即,三种主要的HLA型决定簇基因进行编辑能够产生对于许多受体都是免疫相容的用于移植的细胞,以及包括该细胞的细胞群,从而完成了本发明。
解决问题的方案
本发明的一个目的是提供一种用于产生免疫相容性细胞或细胞群的方法,所述方法包括:在分离的细胞中,通过基因缺失或基因修饰来编辑一种或多种免疫相容性抗原基因的一个或两个等位基因,其中,所述分离的细胞包括选自人类白细胞抗原(HLA)-A、HLA-B和HLA-DR中的至少一种免疫相容性抗原基因。
本发明的另一目的是提供过一种产生免疫相容性细胞或细胞群的方法,所述方法包括:在分离细胞中,通过基因缺失或基因修饰来编辑一种或多种免疫相容性抗原基因的一个或两个等位基因,其中,所述免疫相容性抗原基因中的至少一种选自人类白细胞抗原(HLA)-A和HLA-B和HLA-D并且具有杂合基因型。
本发明的另一目的是提供通过上述产生方法产生的免疫相容性细胞。
本发明的另一目的是提供一种细胞群,所述细胞群包括通过上述产生方法产生的免疫相容性细胞。
本发明的另一目的是提供用于产生免疫相容性细胞或细胞群的方法,所述方法包括:(a)在分离的细胞中,通过基因缺失或基因修饰来编辑一种或多种免疫相容性抗原基因的一个或两个等位基因,其中,所述免疫相容性抗原基因中的至少一种选自人类白细胞抗原(HLA)-A、HLA-B和HLA-DR并且具有杂合基因型;以及(b)收集在步骤(a)中产生的细胞。
本发明的有益效果
根据本发明,通过编辑关键组织相容性抗原(HLA)基因能够产生对许多受体免疫相容的细胞,上述关键组织相容性抗原(HLA)基因可引起对用于移植的异源细胞的免疫排斥。因此,在通过使用该方法构建几种不同的细胞并进行排序时,能够为在需要时可立即用于同种异源移植的细胞疗法提供非常重要的基础。
附图说明
图1为示意图,该示意图示出了通过敲除HLA-A基因和HLA-B基因中一个或两个父源和母源等位基因以及敲除HLA-DRB1基因中两个等位基因中的两个来产生免疫相容性细胞并且构建细胞群的方法。
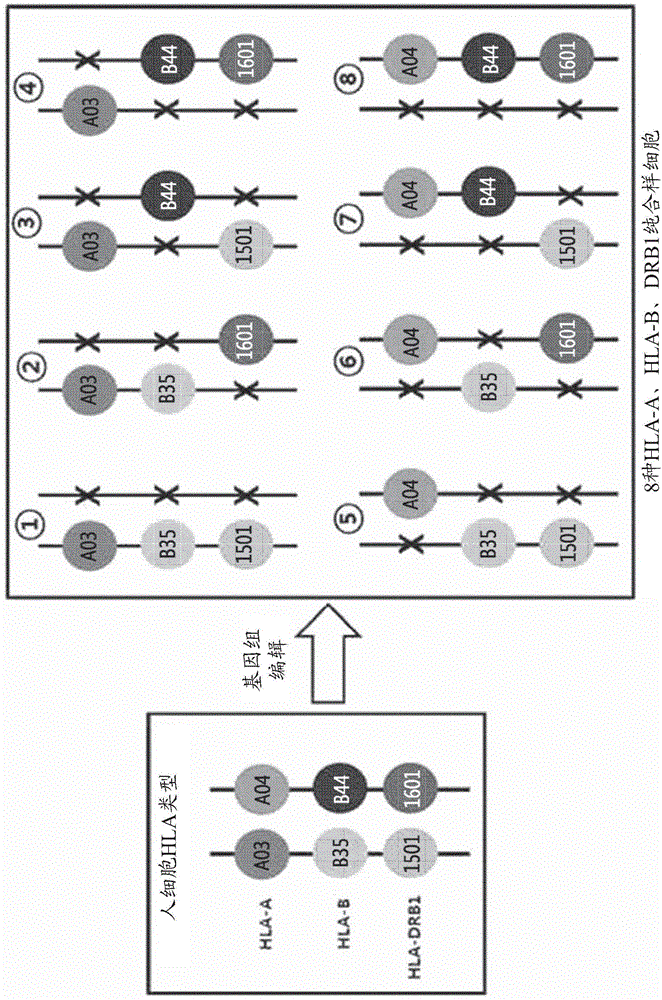
图2为示例图,该示例图示出了在敲除HLA-A基因、HLA-B基因和HLA-DRB1基因的父源和母源等位基因中的一个时,能够由供体的细胞产生的相对于HLA-A基因、HLA-B基因和HLA-DRB1基因为纯合样的总共8种类型的细胞。
图3为示例图,该示例图示出了在敲除HLA-A基因和HLA-B基因的父源和母源等位基因中的一个且敲除HLA-DRB1基因的所述两个等位基因中的两个时,能够由供体的细胞产生的相对于HLA-A基因、HLA-B基因和HLA-DRB1基因为纯合样的总共4种类型的细胞。
图4示出了涉及以下的分析结果:在表达Cas9蛋白和DRB1向导RNA的质粒经电穿孔引入到人类诱导多能干细胞#12之后,在DRB1基因的靶序列的位点中是否诱导插入或缺失(indel)。
图5示出了涉及以下的分析结果:在Cas9蛋白和DRB1向导RNA经电穿孔引入到人类诱导多能干细胞#8之后,在DRB1基因的靶序列的位点中是否诱导插入或缺失。
图6示出了通过序列分析鉴定在图5鉴定的克隆体中克隆体#8的等位基因2中是否存在插入或缺失的结果。
图7示出了涉及以下的分析结果:在Cas9蛋白和DRB1向导RNA经电穿孔引入到人类诱导多能干细胞#12之后,在DRB1基因的靶序列的位点中是否诱导插入或缺失。
图8示出了通过序列分析鉴定在图7鉴定的克隆体中克隆体#4的等位基因1中是否存在插入或缺失的结果。
图9示出了涉及以下的鉴定结果:在Cas9蛋白和DRB1向导RNA经电穿孔引入到人类胚胎干细胞H9#85和CHA15#34以及人类诱导多能干细胞hiPSC12#13之后,在DRB1基因的靶序列的位点中是否诱导插入或缺失。
图10示出了在图9鉴定的克隆体的多能性的鉴定结果。
图11为示意图,示出了利用Cas9蛋白和HLA-A sgRNA敲除细胞中HLA-A基因的一个等位基因的方法,其中,所述细胞DRB1基因的两个等位基因已被敲除。
图12示出了HLA-A的基因型以及A#69克隆体和A#300克隆体的序列的分析结果。
图13为示意图,示出了在图12中鉴定的克隆体中,利用Cas9蛋白和HLA-B sgRNA敲除HLA-B基因的一个等位基因的方法。
图14示出了#53克隆体、#34克隆体、#98克隆体和#134克隆体的HLA-B基因型的分析结果。
图15示出了#53克隆体、#34克隆体、#98克隆体和#134克隆体的HLA-B序列的分析结果。
图16示出了H9克隆体的多能性的鉴定结果,所述H9克隆体具有HLA-A*02+/*03-和HLA-B*35+/*44-基因型,并且其中,DRB1基因的两个等位基因已被敲除。
具体实施方式
在一个实施方式中,本发明提供了一种用于产生免疫相容性细胞的方法,所述方法包括:在分离的细胞中,通过基因缺失或基因修饰来编辑一种或多种免疫相容性抗原基因的一个或两个等位基因,其中,所述分离的细胞包括选自人类白细胞抗原(HLA)-A、HLA-B和HLA-DR的至少一种免疫相容性抗原基因。所述分离细胞可包括杂合的免疫相容性抗原基因或纯合的免疫相容性抗原基因,并且上述编辑步骤可对两个等位基因进行编辑。
另一实施方式,本发明提供了一种用于产生免疫相容性细胞的方法,该方法包括:在分离的细胞中,通过基因缺失或基因修饰来编辑一种或多种免疫相容性抗原基因的一个或两个等位基因,其中,该免疫相容性抗原基因中的至少一种选自人类白细胞抗原(HLA)-A、HLA-B和HLA-DR并且具有杂合基因型。同时,上述产生方法可为用于产生包括上述免疫相容性细胞的细胞群的方法。
此外,上述产生方法可为用于产生免疫相容性细胞的方法,该方法包括:在分离的细胞中,通过基因缺失或基因修饰来编辑免疫相容性抗原基因的一个或两个等位基因,其中,免疫相容性抗原基因(HLA-A或HLA-B)具有杂合基因型。如上所述,该产生方法可为用于产生包括该免疫相容性细胞的细胞群的方法。然而,但并不特别限于此。
为了用于治疗目的,所有细胞疗法必须克服的最重要的障碍是免疫排斥。引起免疫排斥的本质是被称为主要组织相容性抗原(MHC)的6种HLA(HLA-A、HLA-B、HLA-C、HLA-DP、HLA-DQ和HLA-DR)细胞表面蛋白。已知的是,人类表达源自父源基因的六个基因和源自母源基因的六个基因,即,共计6对。更具体地,一般体细胞总共仅表达3对,即,属于MHC I类的HLA-A、HLA-B和HLA-C,而免疫细胞总共表达6对,MHC I类和MHC II类的总和。
HLA表面抗原的作用是在细胞表面上展示细胞中存在的蛋白片段,并且使得通过免疫细胞能够检测到在体内可能发生的感染或突变。为此,它们可被称为抗原呈递蛋白。
当进行细胞疗法或组织移植(包括骨髓移植)时,如果这些抗原呈递蛋白病不是自体的,则它们成为移植受体体内存在的免疫细胞的主要靶标。这是因为每个人具有在相应HLA基因上的多个遗传多态性。例如,由于供体的HLA-A基因不同于受体的HLA-A基因,受体体内的免疫细胞识别到了差异并且攻击供体细胞。最终,由于免疫排斥而导致移植的失败。该现象适用于所有的HLA-A基因,以及HLA-B基因、HLA-C基因、HLA-DP基因、HLA-DQ基因和HLA-DR基因。根据迄今为止所积累的临床经验,已知的是甚至通过在多种多态性中匹配3对HLA-A、HLA-B和HLA-DRB1(或四对,包括至多HLA-C),而不需匹配所有的六对HLA基因(MHC I类和II类)就能够确保相当高的移植成功率。
就该点而言,本发明的特征在于提供一种能够产生免疫相容性细胞的方法,其中,免疫相容性抗原通过基因缺失或基因修饰具有纯合样基因型或半合基因型。
在本发明中,术语“免疫相容性抗原纯合性”是指选自来自供体父源和母源地遗传的HLA-A基因、HLA-B基因和HLA-DR基因的一种、两种或三种基因型具有完全相同的HLA基因型。具体地,免疫相容性抗原纯合性可包括各个HLA-A基因和HLA-B基因完全相同的基因型,但并不限于此。此外,在包括HLA-DR基因时,免疫相容性抗原纯合性可包括各个HLA-A基因、HLA-B基因和HLA-DR基因完全相同的基因型,但并不限于此。
对于更具体的非限制性实施例,源自于供体(父源HLA基因型和母源HLA基因型是相同的)的细胞具有[HLA-A*11(在下文中,省略HLA)、B*51、DRB1*16(父源)/A*11、B*51、DRB1*16(母源)],并且可为具有免疫相容性抗原的纯合体。在该情况下,其移植可适用于如下HLA基因型所有组合的受体:在六对中仅三对是相同的,诸如[A*11、B*51、DRB1*16(父源)/A*24、B*34、DRB1*08(母源)]的受体,[A*11、B*15、DRB1*04(父源)/A*24、B*51、DRB1*16(母源)]的受体、[A*03、B*08、DRB1*16(父源)/A*11、B*51、DRB1*09(母源)]的受体,或者[A*02、B*51、DRB1*07(父源)/A*11、B*44、DRB1*16(母源)]的受体。存在这样的报道:对于超过90%的人免疫相容性细胞疗法,需要携带免疫相容性纯合抗原的约200个细胞系(Taylor C,Banking on human embryonic stem cells:estimating the number of donor cell lines needed for HLA matching,2005,Lancet,366:2019-2025)。因此,在通过HLA基因型筛选找到具有彼此不同的免疫相容性抗原纯合性的200个供体时,能够预先确保能够已知超过90%的免疫相容性细胞系,但是最大的难题在于对这么多人进行HLA基因筛选。
在解决上述问题的一个方面中,本发明人开发了一种利用被称为基因编辑、基因组编辑或基因组工程的方法来产生纯合样细胞的方法。
在本发明中,术语“纯合样”是指通过敲除或通过敲入一对具体杂合的等位基因中的一个等位基因使其仅具有一个等位基因。为了本发明的目的,纯合样是指选自由HLA-A、HLA-B和HLA-DR所组成的组的一种基因、两种基因或三种基因仅具有一个等位基因的状态。更特别地,在本发明中,纯合样是指HLA-A基因、HLA-B基因以及可选地HLA-DR基因仅具有一个等位基因的状态,但并不限于此。
在本发明中,“免疫相容性细胞”可为其中免疫相容性抗原,特别是选自由HLA-A基因、HLA-B基因和HLA-DR基因的一个、两个或三个等位基因以纯合样存在的细胞,但并不特别限于此。更特别地,可为HLA-A基因、HLA-B基因以及可选的HLA-DR基因可以纯合样存在的细胞。
此外,在免疫相容性细胞的HLA-DR基因中,两个等位基因可能已经被完全移除(敲除),但并不限于此。在该情况下,在细胞中,通过基因编辑来缺失或修饰HLA-A基因或HLA-B基因之一的一个等位基因能够产生免疫相容性细胞,其中,在细胞中,HLA-DR基因中的一对等位基因已经被敲除或敲入分离的细胞,其中,HLA-A基因或HLA-B基因中的一种或两种是杂合的。可替代地,在分离的细胞中,HLA-A基因或HLA-B基因中的一种或两种是杂合的,通过基因编辑来缺失或修饰每种HLA-A基因或HLA-B基因的一个等位基因,随后缺失一对HLA-DR基因。此外,在本发明中,在免疫相容性细胞中,选自HLA-A、HLA-B和HLA-DR的一种或多种基因的两个等位基因可本完全移除(敲除)。然而,上述实施例并不用于限定本发明。
通过上文,能够得到可被移植到受体的细胞,该细胞具有在6对中仅两对或一对HLA基因型相同的组合。
此外,根据本发明的方法,当通过基因编辑来缺失或修饰主要组织相容性抗原基因的一个(父源或母源)等位基因时,能够得到如上所述的遗传上纯合样细胞。因此,当收集由此得到细胞(相应等位基因被逐个敲除或敲入)时,能够由一个受体的细胞得到呈现纯合效果的几种类型的细胞(参见图1至图3)。因此,能够提供巨大的优势:得到能够移植至90%人群中的超过200免疫相容性细胞。
在本发明的一个特定实施方式中,免疫相容性细胞可为在其中通过基因缺失或基因修饰已经对杂合基因的一个或两个等位基因进行了编辑的那些细胞。具体地,可通过敲除来进行上述缺失,并且可通过敲入来进行上述修饰,但是并不限于此。
基因组编辑技术/基因编辑技术是能够将靶向定点突变引入到包括人类细胞的动物细胞和植物细胞的基因组序列中,甚至能够在不产生蛋白的非编码DNA序列中敲除或敲入特定基因或引入突变的技术。本发明的方法能够通过基因组编辑技术或基因编辑技术来产生免疫相容性细胞。
在本发明的一个特定实施方式中,基因编辑技术的特征在于利用靶特异性核酸酶。
在本发明中,术语“靶特异性核酸酶”可涉及能够识别和裂解感兴趣的基因组上DNA的特定位置的核酸酶。该核酸酶可包括这样的核酸酶:其中识别基因组上特异性靶序列的结构域以及裂解该特异性靶序列的结构域已经发生融合。核酸酶的实例可包括但不限于:大范围核酸酶;或者工程化核酸酶,尤其是转录激活样效应因子核酸酶(TALEN),该酶为已融合识别基因组上特异性靶序列的结构域和裂解结构域,其中,转录激活样(TAL)效应因子源自植物致病基因;锌指核酸酶;或源自于CRISPR微生物免疫系统的RGEN(RNA-向导的工程化核酸酶)。相对于本发明的目的,利用RGEN的方法是简单的并且能够实现更加令人满意的结果,但是并不意味着特别限于此。此外,相对于本发明的目的,前述基因编辑可利用HLA-A特异性核酸酶、HLA-B特异性核酸酶或HLA-DR特异性核酸酶来进行,并且该核酸酶优选地为工程化核酸酶,但并不限于此。
在利用核酸酶,特别是工程化核酸酶进行敲除过程或敲入过程时,与不需要使用除核酸酶之外的供体DNA的敲除过程不同,敲入过程同时使用供体DNA和核酸酶。供体DNA是指这样的DNA:该DNA包括通过核酸酶引入到裂解的染色体上的位置处的基因。供体DNA可包括用于重组的左侧翼臂和右侧翼臂。此外,供体DNA可任选地包括选择标记物,但并不限于此。
靶特异性核酸酶识别包括人类细胞的动物细胞和植物细胞的基因组中的特定核苷酸序列,从而引起双链断裂(DSB)。DSB通过细胞内的同源重组或非同源末端连接(NHEJ)机制来有效修复。在该过程中,可以将目标突变引入到靶位置。靶特异性核酸酶可以是人工的或工程化的和非天然存在的。
核酸酶可为锌指核酸酶(ZFE)。
ZFN包括选择的基因和锌指蛋白,该锌指蛋白改造为与切割结构域或切割半结构域中的靶位点结合。ZFN可为包括锌指DNA结合结构域和DNA切割结构域的人工的限制酶。本文中,锌指DNA结合结构域可改造为与所选序列结合的结构域。例如,Beerli et al.(2002)Nature Biotechnol.20:135-141;Pabo et al.(2001)Ann.Rev.Biochem.70:313-340;Isalan et al,(2001)Nature Biotechnol.19:656-660;Segal et al.(2001)Curr.Opin.Biotechnol.12:632-637;以及Choo et al.(2000)Curr.Opin.Struct.Biol.10:411-416可通过引入而并入本文。与天然存在的锌指蛋白相比,改造的锌指结合结构域可以具有新的结合特异性。改造方法包括各种类型的合理设计和选择,但并不限于此。合理设计包括使用包含例如三重(或四重)核苷酸序列以及单个锌指氨基酸序列的数据库。此时,各个三重或四重核苷酸序列与结合至特定三重或四重序列的一个或多个锌指序列组合。
靶序列的选择,融合蛋白(以及编码它们的多核苷酸)的设计和构建是本领域技术人员所已知的,并且详细描述在美国专利2005/0064474和2006/0188987,在此上述文献通过引用整体并入本文。此外,如这些参考文献以及相关领域中的其他参考文献所公开的,锌指结构域和/或多锌指蛋白可以通过任何合适的接头序列连接在一起,例如,长度为五个或更多个氨基酸的接头。美国专利号6479626、6903185和7153949中公开了长度为六个或更多个氨基酸的接头序列的实例。本文所述的蛋白可以包括蛋白的每个锌指之间的合适接头的任何组合。
此外,核酸酶,诸如ZFN包括核酸酶活性部分(即切割结构域和切割半结构域)。正如所已知的,例如,在不同于锌指DNA结合结构域的核酸酶的切割结构域中,切割结构域可以与DNA结合结构域是异源的。异源的切割结构域可以源自于任何核酸内切核酸酶或外切核酸酶。切割结构域可源自于的示例性内切核酸酶可包括限制性内切核酸酶和大范围核酸酶,但并不限于此。
类似地,切割半结构域可为源自于任何如上所述的需要用于切割活性的二聚化的核酸酶或其部分。在融合蛋白包括切割半结构域时,通常需要两个融合蛋白用于切割。可替代地,可使用包括两个切割半结构域的单一蛋白。两个切割半结构域也可以源自于同一内切核酸酶(或其功能片段),或者每个切割半结构域可源自于不同的内切核酸酶(或其功能片段)。此外,优选的是,布置两个融合蛋白的靶位点,以通过两个融合蛋白结合至其靶位点,使得半切割结构域相对于彼此空间取向,由此切割半结构域通过二聚化形成功能切割结构域。因此,在一个实施方式中,靶位点的相邻边缘间隔5个至8个核苷酸,或者间隔15个至18个核苷酸。然而,在两个靶位点之间可插入任何整数数目的核苷酸或核苷酸对(例如,2个至50个核苷酸对或更多)。通常,切割位点位于靶位点之间。
限制性内切核酸酶(限制性酶)存在于许多物种中,并且它们可以序列特异性结合DNA(在靶位点),从而在结合位点处或结合位点附近切割DNA。一些限制性酶(例如,IIS型)在移除识别位点的位点处切割DNA,并且包括可分离的结合结构域和可切割结构域。例如,IIS型酶FokI催化从一条链上的识别位点移除9个核苷酸以及在其余的一条链上从识别位点移除13个核苷酸的DNA的双链切割。因此,在一个实施方式中,融合蛋白包括来自IIS型限制性酶的至少一个切割结构域(或切割半结构域)和一个或多个锌指结构域(其可为改造的或者可以非改造的)。
在本发明中,术语“TALEN”是指能够识别和切割DNA的靶区域的核酸酶。TALEN是指包括TALE结构域和核苷酸切割结构域的融合蛋白。在本发明中,术语“TAL效应因子核酸酶”和“TALEN”可以互换使用。已知的是,在各种植物物种被黄单胞属(Xanthomonas)细菌感染时,TAL效应因子是由它们的III型分泌系统所分泌的蛋白。蛋白可以与宿主植物中的启动子序列相关,从而激活有助于细菌感染的植物基因的表达。该蛋白通过中心重复结构域识别植物DNA序列,上述中心重复结构域由34个或更少个各种数目的氨基酸重复组成。因此,据认为,TALE可为用作基因组工程中的工具的新平台。然而,为了产生具有基因组编辑活性的功能性TALEN,迄今为止未知的几个关键参数必须限定如下:i)TALE的最小DNA结合结构域,ii)形成一个靶区域的两个半位点之间的间隔区的长度,以及iii)使FokI核酸酶结构域连接到dTALE的连接子或融合连接。
在本发明中,TALE结构域是指通过一个或多个TALE-重复模块以序列特异性方式结合至核苷酸的蛋白质结构域。TALE结构域包括至少一个TALE重复模块,更具体地1至30个TALE重复模块,但不限于此。在本发明中,术语“TAL效应引子结构域”和“TALE结构域”可互换使用。TALE结构域可以包括半个TALE重复模块。关于TALEN,国际专利WO/2012/093833或美国专利公开号2013-0217131中所公开的全部内容通过引用并入本文。
在本发明中,术语“RGEN”是指包括靶DNA特异性的向导RNA和Cas蛋白作为组分的核酸酶。也就是说,例如,本发明中的RGEN可包括和Cas蛋白以及与HLA-A基因、HLA-B基因或HLA-DR基因的特定序列特异性结合的向导RNA,但不限于此。
在本发明中,RGEN可以以靶DNA特异性的向导RNA或编码向导RNA的DNA的形式;以及分离的Cas蛋白或编码Cas蛋白的核酸的形式应用于细胞,但不限于此。
在本发明的更具体的实施方式中,RGEN可以以下的方式应用于细胞:1)靶DNA特异性的向导RNA和分离的Cas蛋白的形式,以及2)编码向导RNA的DNA和编码Cas蛋白的核酸。
RGEN以上述1)形式转移到细胞被称为“RNP递送”,但不限于此。
在以分离的向导RNA的形式应用时,向导RNA可以在体外转录,但不限于此。
另外,编码向导RNA的DNA和编码Cas蛋白的核酸可用作分离的核酸本身,并且可以以包括表达盒的载体形式存在,该表达盒用于表达向导RNA和/或Cas蛋白,但不限于此。
载体可为病毒载体、质粒载体或农杆菌(Agrobacterium)载体,病毒载体的类型可包括AAV(腺相关病毒),但不限于此。
编码向导RNA的DNA和编码Cas蛋白的核酸分别存在于各个载体中,或者可存在于同一载体中,但不限于此。
即使对于本说明书中描述的更具体的实施方式,也可完全应用每一个上述实施方式。
在本发明中,RGEN可包括与HLA-A基因、HLA-B基因或HLA-DR基因的特定序列特异性结合的向导RNA或编码该向导RNA的DNA,以及Cas蛋白或编码该Cas蛋白的核酸序列,但不限于此。在本发明中,术语“Cas蛋白”是CRISPR/Cas系统的主要蛋白组分,并且为能够形成活化的内切核酸酶或切口酶的蛋白。
Cas蛋白可以与crRNA(CRISPR RNA)和tracrRNA(反式激活crRNA)形成复合体,从而显示其活性。
可从已知的数据库,例如NCBI的GenBank(国家生物技术信息中心)获得Cas蛋白或其上的遗传信息。更具体地,Cas蛋白可为Cas9蛋白。此外,Cas蛋白可为源自于链球菌(Streptococcus)属的蛋白,尤其为源自于酿脓链球菌(Streptococcus pyogens)的Cas蛋白,并且更尤其为Cas9蛋白。此外,Cas蛋白可为源自于弯曲杆菌(Campylobacter)属的蛋白,尤其为源自于空肠弯曲杆菌(Campylobacter jejuni)的Cas蛋白,并且更尤其为Cas9蛋白。然而,本发明并不限于上述实施例。
此外,除了天然存在的蛋白之外,本发明所使用的Cas蛋白包括:与向导RNA协同激活的内切核酸酶或其能够作为切口酶的变体。Cas9蛋白的变体可为Cas9的突变蛋白,其中催化性天冬氨酸残基变为任何其他氨基酸。具体地,其他氨基酸可为丙氨酸,但不限于此。
在本发明中,Cas蛋白可为重组蛋白。
在细胞、核酸、蛋白或载体的背景下使用时,术语“重组”是指例如在其中引入异源核酸或蛋白的修饰的细胞、核酸、蛋白或载体,经修饰的天然核酸酸或蛋白,或源自于修饰的细胞的细胞。因此,例如,利用人密码子表通过重建编码Cas蛋白的氨基酸序列可产生重组Cas蛋白。
Cas蛋白或编码该Cas蛋白的核酸可以是其中Cas蛋白能够在细胞核中起作用的形式。
分离的Cas蛋白也可以为易于引入细胞的形式。作为实例,Cas蛋白可以连接到细胞穿透肽或蛋白转导结构域。蛋白转导结构域可为聚精氨酸或HIV衍生的TAT蛋白,但不限于此。本领域众所周知的是,除了上述实施例之外,还存在各种类型的细胞穿透肽或蛋白质转导结构域,因此本领域技术人员可以将各种实施例应用于本发明而不受限于上文。
此外,编码Cas蛋白的核酸可进一步包括核定位信号(NLS)序列。因此,包括编码Cas蛋白的核酸的表达盒可包括NLS序列以及调节序列,诸如用于表达Cas蛋白的启动子序列。然而,本发明并不限于此。
Cas蛋白可连有便于分离和/或纯化的标签。作为实例,根据需要,可连有诸如His标签、Flag标签或S标签、或者GST(谷胱甘肽S-转移酶)标签、MBP(麦芽糖结合蛋白)标签等小肽标签,但并不限于此。
在本发明中,术语“向导RNA”是指特异性结合特定靶序列的靶DNA特异性RNA,并且其结合Cas蛋白,从而可以将Cas蛋白导向至靶DNA。
在本发明中,向导RNA可为包括两种RNA,即,作为组分的crRNA(CRISPR RNA)和tracrRNA(反式激活crRNA)的双重RNA;或者为包括第一区域和第二区域的形式,所述第一区域包括与靶DNA序列的互补序列进行碱基配对的序列,所述第二区域包括与Cas蛋白相互作用的序列,向导RNA更尤其是sgRNA(单链RNA),在sgRNA中,crRNA和tracrRNA的主要部分已融合。
sgRNA可包括:具有能够与靶DNA序列的互补序列进行碱基配对的序列的部分(也可以称为间隔区、靶DNA识别序列、碱基配对区等),以及用于Cas蛋白结合的发夹结构。更具体地,sgRNA可包括:具有能够与靶DNA序列的互补序列进行碱基配对的序列的部分;用于Cas蛋白结合的发夹结构;以及终止子序列。上述结构可以以5'至3'的顺序依次存在。然而,并不限于此。
如果向导RNA包括crRNA和tracrRNA的必要部分以及能够与靶标互补地进行碱基配对的部分,则任何向导RNA均可用于本发明。
crRNA可与靶DNA杂交。
RGEN由Cas蛋白和双重RNA组成;或者可由Cas蛋白和sgRNA组成。此外,RGEN可包括编码Cas蛋白的核酸和编码双重RNA的核酸作为组分;或者包括编码Cas蛋白的核酸和编码sgRNA的核酸,但并不限于此。
向导RNA,尤其是crRNA或sgRNA可包括能够与靶DNA序列的互补序列进行碱基配对的序列,并且在crRNA或sgRNA的上游部分,尤其是在crRNA或sgRNA或5'端进一步包括一个或多个额外的核苷酸或双重RNA。额外的核苷酸可为鸟嘌呤(G),但并不限于此。
更具体地,本发明的基因编辑可通过将向导RNA或编码该向导RNA的DNA,以及编码Cas蛋白的核酸或Cas蛋白本身引入到细胞中来进行,其中所述向导RNA与HLA-A基因、HLA-B基因或HLA-DR基因的特定序列特异性结合。也就是说,通过使用如前所述的CRISPR/Cas系统(一种核酸酶)的RGEN方法移除主要组织相容性抗原基因中的一个(父源或母源)等位基因,从而使得仅剩余一个等位基因。最后,可以构建具有基因纯合样效应的细胞。
可被用于通过RGEN方法敲除HLA-DRB1基因的等位基因的靶序列可包括通常存在于所有人类中的保守序列,例如,存在于4HLA-DRB1中的:5'-ATCCAGGCAGCATTGAAGTCAGG-3'(SEQ ID NO:1)、5'-CCAGGCAGCATTGAAGTCAGGTG-3'(SEQ ID NO:2)、5'-CCTTCCAGACCCTGGTGATGCTG-3'(SEQ ID NO:3)或5'-CCAGACCCTGGTGATGCTGGAAA-3'(SEQ ID NO:4),但并不限于此。
可被用于通过RGEN方法敲除HLA-A基因的等位基因的靶序列可包括,例如存在于HLA-A外显子4中的:5'-CCCTGCGGAGATCACACTGACCT-3'(SEQ ID NO:5)、5-CCTGCGGAGATCACACTGACCTG-3'(SEQ ID NO:6)、5'-GAGACCAGGCCTGCAGGGGATGG-3'(SEQ ID NO:7)或5'-CACCTGCCATGTGCAGCATGAGG-3'(SEQ ID NO:8),但并不限于此。
可被用于通过RGEN方法敲除HLA-B基因的等位基因的靶序列可包括,例如存在于HLA-B外显子4中的:5'-ACCCTGAGGTGCTGGGCCCTGGG-3'(SEQ ID NO:9)、5'-GATCACACTGACCTGGCAGCGGG-3'(SEQ ID NO:10)、5'-ACACTGACCTGGCAGCGGGATGG-3'(SEQ ID NO:11)、5'-GACCTGGCAGCGGGATGGCGAGG-3'(SEQ ID NO:12)或5'-CCTTCTGGAGAAGAGCAGAGATA-3'(SEQ ID NO:13),但并不限于此。
在本发明的一个实施方式中,使用了如下的方法:对转移至细胞中的Cas 9蛋白和识别特定HLA基因靶序列sgRNA进行编辑以进行HLA基因的等位基因敲除。
具体来说,上述方法可为:(1)在细菌中过表达Cas9蛋白并对其进行纯化,以及体外产生识别特异性HLA靶序列的sgRNA(单向导RNA),随后将Cas蛋白和sgRNA转移到细胞中的方法;或者(2)将表达Cas9蛋白和sgRNA的质粒DNA转染到细胞中并在其中进行表达的方法,但并不限于此。
此外,根据本发明,在将蛋白质、RNA或质粒DNA转移到细胞的方法中,可使用本领域已知的各种方法,诸如电穿孔、脂质体、病毒载体、纳米颗粒和蛋白易位结构域(PTD)融合蛋白方法,但并不限于此。
此外,本发明的方法可应用于去分化干细胞(诱导多能干细胞)、胚胎干细胞以及所有细胞。也就是说,作为一项技术,其可应用于各种细胞是有利的。
该方法可应用于所有细胞,即干细胞(诱导多能干细胞、胚胎干细胞、体细胞核转移衍生的胚胎干细胞和成体干细胞)和体细胞。此外,细胞来源于人,但并不限于此。
本文所提及的成体干细胞包括可从人胚胎、新生儿和成体中获得的所有成体干细胞,以及脐带血干细胞,胎盘干细胞,脐带胶质干细胞,羊水干细胞,羊膜上皮细胞,胚外干细胞和源自于他们的经遗传修饰的细胞。
此外,本文所提及的体细胞包括可从胚胎以及新生儿和成体中获得的所有细胞,以及源自于他们的所有经遗传修饰的细胞。
在HLA-A、HLA-B和HLA-DR中的两种或更多种基因具有杂合基因型时,或在HLA-A基因和HLA-B基因具有杂合基因型时,根据本发明产生免疫相容性细胞的方法可通过基因编辑来依次或同时(并且尤其可依次进行)缺失或修饰来自相应细胞的杂合基因的等位基因,但并不限于此。此外,在HLA-A基因、HLA-B基因和HLA-DR基因都具有杂合基因型时,或者在HLA-A基因和HLA-B基因具有杂合基因型时,该产生方法通过基因编辑可缺失或修饰来自分离细胞的相应基因的一个或两个等位基因,并且通过基因编辑可依次或同时缺失或修饰相应基因的等位基因。此外,该产生方法可包括以下步骤:通过基因编辑移除分离细胞中的HLA-DR基因的一对等位基因,在分离细胞中,HLA-A基因、HLA-B基因和HLA-DR基因都具有杂合基因型;以及,通过基因编辑移除或修饰HLA-A基因和HLA-B基因中每者中的一个或两个等位基因,但并不限于此。由于DRB1作为MHC II类蛋白之一仅在一些细胞(诸如B细胞)中表达,因此具有能够进行双链体敲除并且简化HLA组合的类型的优点,但是并不限于此。根据该方法,在一种类型的细胞能够产生四种不同的纯合样细胞。
在本发明的一个特定实施方式中,对胚胎干细胞和诱导多能干细胞进行双等位基因敲除,以阻断Drb1基因(图9);并且随后,通过基因编辑移除HLA-A基因和HLA-B基因中每者中的一个等位基因,从而产生四种不同的纯合样细胞(图14和图15)。
此外,本发明的产生方法可以进一步包括:通过基因编辑移除或修饰选自除了HLA-A基因、HLA-B基因和HLA-DR基因之外的HLA-C基因、HLA-DP基因和HLA-DQ基因的一种或多种基因的等位基因的步骤。显然,对于这种基因编辑,同样适用于上文中所述的内容。
此外,本发明的产生方法可进一步包括:对所产生的细胞的HLA类型进行分析。该步骤可使用本领域已知的用于分析基因型的各种方法来进行。例如,该步骤可通过使用PCR扩增感兴趣的HLA基因的靶序列区域,且随后分析其序列的方法来进行,但并不限于此。
此外,上述方法可进一步包括:从所产生的免疫相容性细胞群中选择待移植的具有HLA型的细胞。该选择步骤中可在分析HLA基因型的步骤之前进行。在分析了HLA基因型后,可通过分析结果选择所需细胞。该过程可以向受体提供合适的HLA类型的细胞。
在本发明的另一实施方式中,提供了通过产生方法产生的免疫相容性细胞。
该产生方法的详细内容如上文所述。
在另一实施方式中,本发明提供了细胞群,所述细胞群包括通过上述产生方法产生的免疫相容性细胞。
该产生方法的详细内容如上文所述。上述细胞群也可被称为细胞库。
在另一实施方式中,本发明提供了用于产生免疫相容性细胞群的方法,该方法包括:(a)在分离的细胞中,通过基因缺失或修饰来编辑免疫相容性抗原基因的一个或两个等位基因,其中,上述免疫相容性抗原基因中的至少一种选自HLA-A、HLA-B和HLA-DRB1并且具有杂合基因型,或者在分离的细胞中,上述免疫相容性抗原基因中的至少一种选自HLA-A和HLA-B且具有杂合基因型;以及,(b)收集步骤(a)中所产生的细胞。
步骤(a)的细节如前文所述。此外,细胞群可被称为细胞库。
在步骤(b)中,通过收集步骤(a)中所产生的细胞能够构建具有各种HLA基因型的细胞群。
此外,在进行步骤(a)之后且在进行步骤(b)之前,本发明的产生方法可进一步包括:(a')对步骤(a)获得的分离的细胞的HLA基因型进行鉴定,从而产生HLA基因型鉴定的细胞群,即细胞群。
本发明的实例
下文,将通过实施例来详细地描述本发明。
然而,这些实施例仅用于说明本发明,而不用于限制本发明。
已知的是,需要匹配的最重要的HLA分子是I类HLA-A、HLA-B以及II类HLA-DR。为了创建HLA-A纯合的人类多能干细胞文库,我们设计了将Cas9靶向Drb1基因、HLA-A基因和HLA-B基因的gRNA,并测试了在ES细胞和iPS细胞中针对定点特异性突变的能力。图1为概述我们的策略的示意图。
材料和方法
细胞培养
在补充有10μM ROCK抑制剂Y27632(Santa Cruz)的E8培养基(Invitrogen)中,使hESC和iPSC在Geltrex(Invitrogen)包被的板上维持24小时。每4天至5天,用EDTA传代细胞。
向导RNA
根据制造商的说明书,使用MEGA shortscript T7试剂盒(Ambion)在体外转录RNA。通过两个互补寡核苷酸的退火和延伸来产生sgRNA的模板。向导RNA序列具有相同的靶DNA序列,并且为23nt序列,其中序列3'末端是“NGG”。
转染
根据制造商的方案,利用Program CB-150通过Amaxa P3原代细胞4D核转染试剂盒转染hESC和iPSC。简而言之,用Cas9蛋白转染2×105个细胞,该Cas9蛋白来自于预混合有体外转录的sgRNA(40μg)的Toolgen(30μg)。使混合有sgRNA的Cas9蛋白溶解在无核酸酶的水中,并在室温下孵育10分钟。将不超过10μL的Cas0-sgRNA混合物加入到100μL的核转染(Nucleofection)溶液中。转染3天后,对细胞进行分析。
T7E1测定
使用DNeasy血液&组织试剂盒(QIAGEN)提取基因组DNA。使用PicoMaxx高保真PCR系统(Agilent)产生包括Cas9靶位点的PCR扩增子。通过加热使PCR扩增子变性,并使用热循环仪退火来形成异源双链DNA,随后在37℃下用T7内切核酸酶1(New England Biolabs)消化20分钟,然后使用琼脂糖凝胶电泳进行分析。对于测序分析,使用TA克隆载体(pGEM-T Easy Vector;Promega)来使PCR产物用于亚克隆。对重建的质粒进行纯化,并且对单个克隆进行测序(Macrogen Inc.)。
碱性磷酸酶染色
根据制造商的说明书,使用碱性磷酸酶染色试剂盒II(Stemgent)进行碱性磷酸酶染色。用4%多聚甲醛固定细胞,用Tris-缓冲盐水/0.05%Tween 20(Sigma)洗涤,并且用AP染色溶液进行染色。
免疫荧光
用4%甲醛固定hESC,并且在室温下,用含0.1%Triton X-100的磷酸盐缓冲盐水(PBS;Invitrogen)通透化30分钟。随后,用含0.03%Triton X-100的PBS(洗涤缓冲液)洗涤细胞。用溶解在洗涤缓冲液中的5%牛血清白蛋白溶液使该固定的样品封闭1小时,随后在4℃下用一抗孵育24小时。用洗涤缓冲液洗涤样品,然后与FITC缀合的二抗(分子探针,Molecular Probes)孵育2小时。用DAPI(Vector Laboratories)对载玻片进行反染色以染色细胞核。
靶向深度测序
使用Phusion聚合酶(New England BioLabs)扩增跨越靶位点和潜在非靶位点的基因组DNA区段。使用Illumina MiSeq对所得PCR扩增产物进行配对末端测序。
核型分析
通过GenDix Inc对人类染色体核型进行分析。
实施例1:通过HLA-A基因、HLA-B基因和HLA-DRB1基因的单等位基因缺失来产生纯合样细胞
在分离的细胞(在所述分离的细胞中,人类白细胞抗原(HLA)-A基因、HLA-B基因和HLA-DR基因具有杂合基因型)中,利用核酸酶通过基因组编辑来进行HLA-A基因、HLA-B基因和HLA-DRB1基因的单等位基因敲除,从而构建其中HLA-A基因、HLA-B基因和HLA-DRB1基因具有纯合样特性的8种细胞。上述过程示于图2的示意图中。
实施例2:通过HLA-A基因和HLA-B基因的单等位基因敲除和HLA-DRB1基因的双等位基因敲除来产生纯合样细胞
在分离的细胞(在所述分离的细胞中,HLA-A基因、HLA-B基因和HLA-DRB1基因具有杂合基因型)中,利用核酸酶通过基因组编辑来进行HLA-DRB1基因的一对等位基因的移除,以及进行HLA基因和HLA-B基因的单等位基因敲除,从而构建其中HLA-A基因和HLA-B基因具有纯合样特性的四种细胞。上述过程示于图3的示意图中。
实施例3:将表达Cas9蛋白和HLA-DRB1特异性向导RNA(DRB1 gRNA)的质粒引入到人类诱导多能干细胞中
通过电穿孔,将表达Cas9蛋白和DRB1gRNA的质粒DNA递送到人类诱导多能干细胞#12(iPSC#12)中。随后,提取源自于iPSC#12的所得菌落的基因组DNA。然后,通过T7内切核酸酶I(T7E1)突变检测测定法分析DRB1基因内的靶序列处是否诱导插入或缺失,并且结果示于图4中。
如图4所示,分析的结果为:在23个iPSC细胞系中,确定有7个iPSC细胞系(红色箭头)在DRB1基因的两个(父源和母源)等位基因诱导了插入或缺失;确定有1个iPSC细胞系(紫色箭头)在等位基因#1(示为等位基因1)处诱导了插入或缺失;以及确定有4个iPSC细胞系(蓝色箭头)在等位基因#2(示为等位基因2)处诱导了插入或缺失。
实施例4:使用电穿孔将Cas9蛋白和HLA-DRB1特异性向导RNA(DRB1gRNA)引入到人类诱导多能干细胞
通过电穿孔,将Cas9蛋白和HLA-DRB1gRNA(非质粒DNA)递送到人类诱导多能干细胞#8(iPSC#8)中。随后,提取源自于iPSC#8的所得菌落的基因组DNA。然后,通过T7内切核酸酶I(T7E1)突变检测测定法分析HLA-DRB1基因内的靶序列处是否诱导插入或缺失,并且结果示于图5中。
如图5所示,分析的结果为:在总共75个iPSC细胞系中,确定有7个iPSC细胞系(红色箭头)在DRB1基因的等位基因#1(示为等位基因1)中诱导了插入或缺失;以及确定有2个iPSC细胞系(蓝色箭头)在等位基因#2(示为等位基因2)上诱导了插入或缺失。图5示出了经分析的总共75个iPSC细胞系中42个细胞系的琼脂糖凝胶分析的结果。
此外,在示出利用人类诱导多能干细胞#8(iPSC#8)诱导插入或缺失的结果的图5中已经看出,克隆体#8已经被鉴定为在等位基因2处包括插入或缺失(参见图5)。为了验证这一点,通过PCR扩增靶序列区,随后进行序列分析。结果示于图6。
因此,如图6所示,证实了等位基因1没有变化,而在等位基因2中插入了一个核苷酸(“A”)。
此外,通过电穿孔,将Cas9蛋白和HLA-DRB1 gRNA(非质粒DNA)递送到人类诱导多能干细胞#12(iPSC#12)中。随后,提取源自于iPSC#12的所得菌落的基因组DNA。然后,通过T7内切核酸酶I(T7E1)突变检测测定法分析HLA-DRB1基因内的靶序列处是否诱导插入或缺失,并且结果示于图7。
如图7所示,分析的结果表明在克隆#4、克隆#6、克隆#9和克隆#10中,在等位基因1中存在插入或缺失,而在等位基因2中没有变化。
在图7中,在克隆#4、克隆#6、克隆#9和克隆#10中,在等位基因1处具有插入或缺失,选择克隆#4,并验证插入或缺失的存在。为此,通过PCR扩增等位基因1和等位基因2的靶序列区域,然后进行序列分析。结果示于图8中。
因此,如图8所示,证实了等位基因1中插入了一个核苷酸(“A”),而等位基因2没有变化。
实施例5:建立完全敲除的Drb1多能干细胞系
II类蛋白主要在B淋巴细胞、巨噬细胞和树突细胞上表达。与I类蛋白不同,它们的损失不会引发NK细胞介导的细胞裂解。因此,我们创建了完全敲除的Drb1 hESC。
为此,使纯化的重组Cas9蛋白与靶向Drb1基因座的体外转录的sgRNA复合。通过电穿孔,将所得蛋白-RNA复合体转染到H9细胞系、CHA15 hES细胞系和hiPSC12细胞系中。在转染72小时后,解离细胞,并在补充有Rho激酶(ROCK)抑制剂的hESC培养基中替换为密度非常低的单细胞。
为了验证Drb1敲除,我们进行了深度测序和Sanger测序。在选定的代表性克隆体H9中,#85显示出5nt和13nt缺失模式,CHA15#34显示出两个等位基因的1nt插入,而在hiPSC12克隆#13中观察到1nt插入和32nt缺失的模式(图9)。我们所有的Drb1敲除克隆体表现出稳定的生长,并且在超过10代呈现圆形紧密压缩形态。
为了进一步验证克隆体的多能性,我们进行了免疫染色。免疫细胞化学分析证实了Drb1 KO克隆适当地表达多能性标记物Oct4、Nanog、AP,并且没有观察到核型异常(图10)。
实施例6:Drb1敲除且HLA-A和HLA-B纯合多能干细胞系的建立
为了得到4个HLA-A和HLA-B纯合细胞系,我们接下来在Dr1b KO克隆中靶向HLA-A和HLA-B。
再次使用Cas9蛋白和体外转录的sgRNA递送方法,我们首先建立HLA-A单等位基因突变细胞。对源自于各克隆的单细胞的测序进行分析以鉴定在靶区域上具有单等位基因插入或缺失图案的克隆。图11总结了源自Drb1KO CHA15 hES细胞系的HLA-A单等位突变克隆体的失活(skim)和突变模式。
等位基因特异性扩增的PCR产物的T7E1分析仅表明在一个等位基因上的特异性切割。测序结果进一步验证了克隆A#69在等位基因A*31上具有1nt插入,而克隆体A#300在等位基因A*33上具有1nt缺失(图12)。
在验证HLA-A突变之后,我们引入了靶向HLA-B基因的sgRNA(图13)。通过等位基因特异性T7E1和测序来检测克隆。图14和图15示出了来自CHA15 hES细胞系的代表性克隆。在H9 hES和iPS12细胞系中同样进行了平行实验。
本发明人未观察到我们的克隆中的任何形态异常或生长异常。最后,我们进一步表征具有Drb1完全KO、HLA-A*02+/*03-和HLA-B*35+/*44-基因型的一个H9克隆体的多能性。免疫细胞化学分析证实了这些细胞表达多能性标记物Oct4和Nanog。
本发明人的研究证实了通过使用CRISPR-Cas9介导的遗传干预能够容易地产生HLA纯合的人类多能干细胞。在本文中,我们得到了这些克隆的小文库,形成预先存在的多能细胞系。因为我们的方案消除了SNT或因子介导的重编程(通常需要进行耗时且昂贵的细胞分离过程以得到供体~受体匹配的细胞系),所以具有重要的意义。本发明人预期我们的方案通过提供与患者免疫匹配的多能干细胞来加速基于细胞的疗法从实验室到临床的过渡。重要的是,使用源自于GMP条件下良好表征的细胞系将显着降低初始成本。此外,基于蛋白的Cas9递送,而不是以质粒形式递送,阻止质粒载体潜在整合到基因组中。
总之,本发明人的研究能够从易得的hES细胞系或iPSC细胞系来生成与患者免疫匹配的多能干细胞。
从上文的描述中,本发明所属领域的技术人员可理解的是,在不改变本发明的技术精神或本质特征的情况下,本发明可以以其他具体形式实施。在这方面,上述实施例应当被理解为是示例性的而不用于在所有方面中进行限制。必须解释的是,除了前文的详细描述外,从所附权利要求及其等同物的含义和范围所衍生的所有改变或改动形式均包括在本发明的范围内。
<110> 车医科学大学校产学协力团
基础科学研究院
<120> 核酸酶介导的编辑编码HLA的基因所产生的免疫相容性细胞
<130> OPA15186-PCT
<150> KR 10-2014-0101203
<151> 2014-08-06
<160> 13
<170> KopatentIn 2.0
<210> 1
<211> 23
<212> DNA
<213> 人工序列
<220>
<223> HLA-DRB1 靶序列
<400> 1
atccaggcag cattgaagtc agg 23
<210> 2
<211> 23
<212> DNA
<213> 人工序列
<220>
<223> HLA-DRB1 靶序列
<400> 2
ccaggcagca ttgaagtcag gtg 23
<210> 3
<211> 23
<212> DNA
<213> 人工序列
<220>
<223> HLA-DRB1 靶序列
<400> 3
ccttccagac cctggtgatg ctg 23
<210> 4
<211> 23
<212> DNA
<213> 人工序列
<220>
<223> HLA-DRB1 靶序列
<400> 4
ccagaccctg gtgatgctgg aaa 23
<210> 5
<211> 23
<212> DNA
<213> 人工序列
<220>
<223> HLA-A 靶序列
<400> 5
ccctgcggag atcacactga cct 23
<210> 6
<211> 23
<212> DNA
<213> 人工序列
<220>
<223> HLA-A 靶序列
<400> 6
cctgcggaga tcacactgac ctg 23
<210> 7
<211> 23
<212> DNA
<213> 人工序列
<220>
<223> HLA-A 靶序列
<400> 7
gagaccaggc ctgcagggga tgg 23
<210> 8
<211> 23
<212> DNA
<213> 人工序列
<220>
<223> HLA-A 靶序列
<400> 8
cacctgccat gtgcagcatg agg 23
<210> 9
<211> 23
<212> DNA
<213> 人工序列
<220>
<223> HLA-B 靶序列
<400> 9
accctgaggt gctgggccct ggg 23
<210> 10
<211> 23
<212> DNA
<213> 人工序列
<220>
<223> HLA-B 靶序列
<400> 10
gatcacactg acctggcagc ggg 23
<210> 11
<211> 23
<212> DNA
<213> 人工序列
<220>
<223> HLA-B 靶序列
<400> 11
acactgacct ggcagcggga tgg 23
<210> 12
<211> 23
<212> DNA
<213> 人工序列
<220>
<223> HLA-B 靶序列
<400> 12
gacctggcag cgggatggcg agg 23
<210> 13
<211> 23
<212> DNA
<213> 人工序列
<220>
<223> HLA-B 靶序列
<400> 13
ccttctggag aagagcagag ata 23
- 还没有人留言评论。精彩留言会获得点赞!