用于高温聚合的齐格勒‑纳塔催化剂的制作方法
本发明涉及用于聚烯烃的高温溶液聚合的镁-钛催化剂。
背景技术:
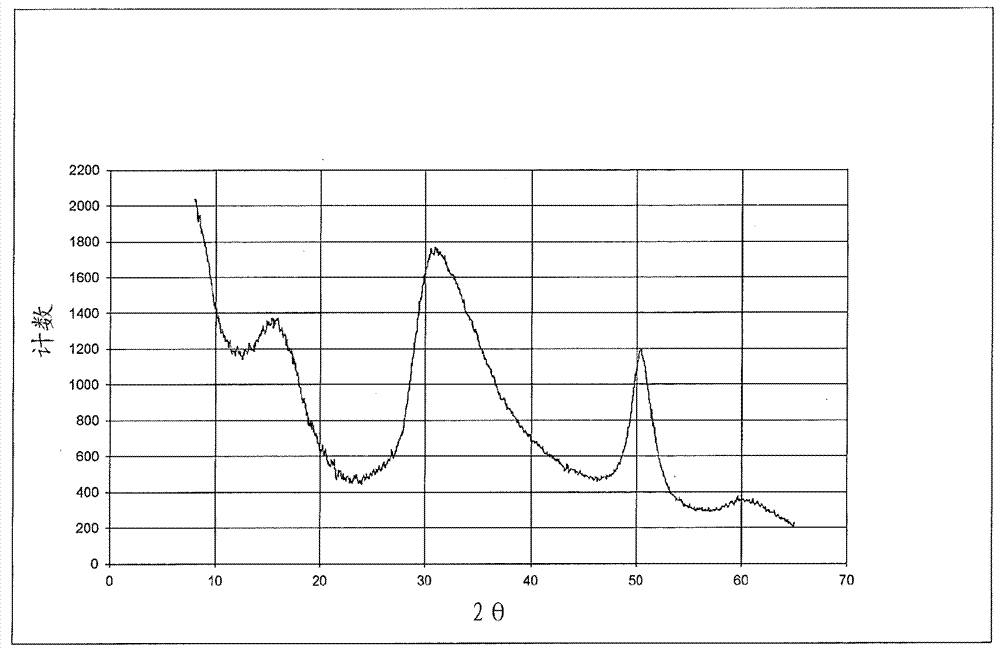
:用于烯烃聚合的镁-钛催化剂在商业上广泛使用。通常而言,这些催化剂包含镁卤化物组分(通常是二氯化镁)和沉积于二氯化镁上的钛组分。所得镁-钛复合物常被称为“主催化剂”,这是由于其需要助催化剂或活化剂来产生高度反应性的聚合催化剂体系。可以首先合成主催化剂,然后在之后添加至聚合反应器。或者,主催化剂可以通过“管线内(in-line)混合技术”(邻近聚合反应器)来制备,并直接添加至反应器。许多原始的齐格勒-纳塔催化剂活性不足以使催化剂残留物能够留在聚合物中而不引起品质问题(例如聚合物颜色、以及在不合需要的短时期内使聚合物降解/氧化的倾向)。相应地,存在对“高活性驻留式(leave-in)”催化剂的需求,其特征在于,其具有较少的可留在成品聚合物中的会引起问题的催化剂残留物。存在对用于聚烯烃的高温溶液聚合的高度活性镁-钛催化剂的需求,其可以在较低的残留钛和卤素杂质的情况下提供增加的共聚单体引入和较高分子量的聚合物材料。技术实现要素:本发明的一些实施方案提供用于乙烯和α-烯烃的聚合的主催化剂,所述主催化剂包含至少0.2%的可通过epr检测的具有1.950的g值的物质。本发明的一些实施方案提供用于乙烯和α-烯烃的聚合的在δ型mgcl2载体上的主催化剂,所述主催化剂包含式ticl3*[[r4]a[r5o]balx3-c]d的ti3+配合物,其中,a为0至1;b为0至1;c=a+b;d为约0.33至约1.0;每个r4和r5独立地选自c1-8烷基;每个x独立地选自卤素;且进一步地其中,存在的总ti中的至少60%呈ti3+氧化态。本发明的其他实施方案提供用来制备包含ti3+配合物的烯烃聚合主催化剂的方法,所述方法包括:a)通过组合以下物质来形成δ型mgcl2物质:i)选自c5-12烷烃的溶剂中的r2mg,和ii)反应性有机氯化物或hcl,其中,每个r独立地选自c2-8烷基,并且其中所添加的cl和mg的摩尔比为约2.0至约3.0;b)向步骤a中制备的所述δ型mgcl2物质,以任意顺序或同时添加r1xalx3-x和四价钛化合物,其中,al/ti摩尔比为约3至约10;或者c)向步骤a中制备的所述δ型mgcl2物质,先添加r1xalx3-x,其次添加四价钛化合物,接着添加r4yalor53-y,其中,当测量仅由r1xalx3-x供应的al时的al/ti摩尔比为约0.7至约2,并且当测量由r4yalor53-y供应的al时的al/ti摩尔比为约1至约2;且进一步地其中,mg/ti摩尔比为约5至约10;x为1或2;y为1或2;每个r1独立地选自c1-8烷基;四价钛化合物选自tir2x3、ti(or3)x3、tix4、和它们的混合物;每个x独立地选自卤素;每个r2独立地选自c1-8烷基和苄基,并且每个r3、r4和r5独立地选自c1-8烷基。本发明的其他实施方案提供通过以下方法制备的包含ti3+配合物的主催化剂产物,所述方法包括:a)通过组合以下物质来形成δ型mgcl2物质:i)选自c5-12烷烃的溶剂中的r2mg,和ii)反应性有机氯化物或hcl,其中,每个r独立地选自c2-8烷基,并且其中所添加的cl和mg的摩尔比为约2.0至约3.0;b)向步骤a中制备的所述δ型mgcl2物质,以任意顺序或同时添加r1xalx3-x和四价钛化合物,其中,al/ti摩尔比为约3至约10;或者c)向步骤a中制备的所述δ型mgcl2物质,先添加r1xalx3-x,其次添加四价钛化合物,接着添加r4yalor53-y,其中,当测量仅由r1xalx3-x供应的al时的al/ti摩尔比为约0.7至约2,并且当测量由r4yalor53-y供应的al时的al/ti摩尔比为约1至约2;并且进一步地其中,mg/ti摩尔比为约5至约10;x为1或2;y为1或2;每个r1独立地选自c1-8烷基;四价钛化合物选自tir2x3、ti(or3)x3、tix4、和它们的混合物;每个x独立地选自卤素;每个r2独立地选自c1-8烷基和苄基,并且每个r3、r4和r5独立地选自c1-8烷基。本发明的其他实施方案提供溶液烯烃聚合方法,其包括:i)向任选具有一个或多个附加反应器的连续搅拌釜式反应器(cstr),添加选自c5-12烷烃的溶剂和用于聚合的在δ型mgcl2载体上的主催化剂,所述主催化剂包含式ticl3*[[r4]a[r5o]balx3-c]d的ti3+配合物,其中,a为0至1;b为0至1;c=a+b;d为0.33至1.0;每个r4和r5独立地选自c1-8烷基;每个x独立地选自卤素;其中,存在的总ti中的至少60%呈ti3+氧化态;ii)向反应器添加乙烯、氢气和任选一种或多种选自c3-8共聚单体的共聚单体;和iii)向反应器以相对于主催化剂的量为约1至约10的摩尔比添加烷基铝活化剂。本发明的其他实施方案提供通过以下聚合方法制备的烯烃聚合产物,所述方法包括:i)向任选具有一个或多个附加反应器的连续搅拌釜式反应器(cstr),添加选自c5-12烷烃的溶剂和用于聚合的在δ型mgcl2载体上的主催化剂,所述主催化剂包含式ticl3*[[r4]a[r5o]balx3-c]d的ti3+配合物,其中,a为0至1;b为0至1;c=a+b;d为0.33至1.0;每个r4和r5独立地选自c1-8烷基;每个x独立地选自卤素;其中,存在的总ti中的至少60%呈ti3+氧化态;ii)向反应器添加乙烯、氢气和任选一种或多种选自c3-8共聚单体的共聚单体;和iii)向反应器以相对于主催化剂的量为约1至约10的摩尔比添加烷基铝活化剂。本发明的其他实施方案提供选自膜、纤维、模塑或热成型的制品、以及管道涂层的塑料制品,其包含通过下述聚合方法制备的烯烃聚合产物,所述方法包括:i)向任选具有一个或多个附加反应器的连续搅拌釜式反应器(cstr),添加选自c5-12烷烃的溶剂和用于聚合的在δ型mgcl2载体上的主催化剂,所述主催化剂包含式ticl3*[[r4]a[r5o]balx3-c]d的ti3+配合物,其中,a为0至1;b为0至1;c=a+b;d为0.33至1.0;每个r4和r5独立地选自c1-8烷基;每个x独立地选自卤素;其中,存在的总ti中的至少60%呈ti3+氧化态;ii)向反应器添加乙烯、氢气和任选一种或多种选自c3-8共聚单体的共聚单体;和iii)向反应器以相对于主催化剂的量为约1至约10的摩尔比添加烷基铝活化剂。附图简述图1示出来自α型mgcl2的典型xrd谱。图2示出使用在本文中公开并要求保护的方法形成的δ型mgcl2的xrd图谱。图3示出产物5的gpc-ft-ir。图4示出催化剂1的epr谱和模拟。图5示出催化剂2b的epr谱和模拟。图6示出对比实施例a的epr谱和模拟。具体实施方式除在操作实施例中或在另有指明之处外,所有在说明书和权利要求书中使用的指代成分的量、反应条件等的数字或表达均要被理解为在所有情况下被术语“约”修饰。相应地,除非有相反指明,在以下说明书和随附的权利要求书中陈述的数值参数是可以根据本发明期望获得的所需特性而变化的近似值。在最低限度上,并且不试图限制将等同原则应用于权利要求的范围,应当至少根据所报道的有效数字的数值并且通过应用普通舍入技术来解释各数值参数。尽管陈述本发明的宽泛范围的数值范围和参数是近似值,但在具体实施例中陈述的数值则尽可能精确地报道。但任何数值都固有地包含一定的由在其各自的测试测量中存在的标准偏差所必然导致的误差。同时,应当理解的是,在本文中引述的任何数值范围旨在包括在其中包含的所有子范围。例如,范围“1至10”旨在包括之间的全部子范围并包括所引述的最小值1和所引述的最大值10;也即是说,具有等于或大于1的最小值和等于或小于10的最大值。由于所公开的数值范围是连续的,其包括在最小值和最大值之间的每个值。除非另有明确指明,在本申请中指定的多个数值范围是近似值。在本文中表达的所有组成范围在实践中限制在总计100%且不超过100%(体积%或重量%)。在组合物中可以存在多种组分的情况下,各组分的最大量的总和可以超过100%,要理解且本领域技术人员容易理解的是,实际使用的组分的量将符合100%的最大值。必须注意的是,如在本文中和在随附的权利要求中所使用的单数形式“一个”、“一种”和“所述”包括复数指代,除非上下文明确地另有规定。除非在本文中定义,在本文中使用的所有技术、科学术语具有与本发明所属领域的普通技术人员所通常理解的相同含义。术语“烷基”、“烷基基团”和“烷基原子团”可以互换使用,并且是指键合至一个或多个其他部分的饱和的单价直链或支链和环状的烃基基团或原子团。例如,烷基可以键合至氧原子从而形成烷氧基,或者作为在金属上的配体的一部分或者作为配体而键合至金属。术语“烷基”由例如以下基团例示:甲基、乙基、正丙基、异丙基、正丁基、叔丁基、正戊基、金刚烷基、环丙基、环丁基、环戊基、环己基、环辛基等。术语“烷烃”是指具有通式cnh(2n+2)的非芳族饱和烃分子,其中n为整数。烷烃例如可被用作溶剂或气体进料。当在术语之前为cx-y(其中x和y为整数)时,所述基团限于在基团内x至y个碳原子,不包括被称为取代基团的任何取代基。例如,c1-5烷基将包括(但不限于)甲基、异丙基、正丁基、叔丁基、环丙基和环戊基,而c1-5烷烃将包括(但不限于)甲烷、乙烷、戊烷、环戊烷等。术语“卤素原子团”或“卤素”或“卤代”可以互换使用,并且是指氟基团(fluoridegroup)、氯基团(chloridegroup)、溴基团(bromidegroup)或碘基团(iodidegroup)。主催化剂在一个实施方案中,在本文中描述的本发明是用于乙烯和α-烯烃的聚合的在δ型的mgcl2载体上的主催化剂,所述主催化剂包含式ticl3*[[r4]a[r5o]balx3-c]d的ti3+配合物,其中,a为0至1;b为0至1;c=a+b;d为0.33至1.0;每个r4和r5独立地选自c1-8烷基;每个x独立地选自卤素;并且其中,存在的总ti中的至少60%呈ti3+氧化态。尽管x可以为任意卤素,但在一些实施方案中,x为br或cl。在其他实施方案中,x为cl。在一些实施方案中,c为0。在其他实施方案中,c为1。在一些实施方案中,a为0并且b为1。在一些实施方案中,a为1且b为0。在一些实施方案中,a为1且b为1。在一些实施方案中,a为0且b为0。在一些实施方案中,每个r5为c1-4烷基。在其他实施方案中,每个r5为乙基。在一些实施方案中,每个r4为c1-4烷基。在其他实施方案中,每个r4为乙基。镁/钛摩尔比将要被镁-钛聚合催化剂领域的技术人员认识到的是,催化剂活性可受到镁/钛摩尔比的影响。对于本发明的催化剂,优选的摩尔mg/ti比为5/1至10/1,即催化剂中每摩尔的ti优选存在5至10摩尔的mg。在一些实施方案中,mg/ti摩尔比为约5至约8。在其他实施方案中,mg/ti比为约6至约8。所需mg/ti摩尔比可以通过根据本文中所描述的方法来制备主催化剂而获得。主催化剂式和在其中包含的元素比率可以使用标准元素分析技术来确定,所述标准元素分析技术包括但不限于经典的“湿法化学”、中子活化、电感耦合等离子质谱(icp-ms)和x射线衍射谱(xrd)。针对钛价态分布,可以使用针对钛价态分布的氧化还原滴定法(参见j.c.w.chien等人,j.polym.sci.parta:polymchem.1989,27,1499-1514)来分析催化剂样品,或针对钛含量分析,基于astm标准e878-01使用紫外(uv)法来分析催化剂样品。在一些实施方案中,存在的总ti中的至少70%呈ti3+氧化态。在其他实施方案中,存在的总ti中的至少80%呈ti3+氧化态。固体齐格勒催化剂的表征可以通过电子顺磁共振谱(epr)来实现,呈氧化态+3的钛原子的部分对其灵敏。g值归属基于出版物j.c.w.chien等人,j.polym.sci.parta:poly.chem.1982,20,2461-2476。在检验在本文中描述的齐格勒催化剂的epr谱和它们相应的模拟谱时,观察到三组epr峰,并将其归属于物质a、b和c。具有1.910、1.898、1.955的g值的物质a据信为其中来自mgcl2的两个氯离子配位至ticl3的物质(该配合物据信类似于在chien(1982)参考文献中归属于物质a的配合物)。物质b由于峰非常宽而难以界定。当物质c以1.950的g值存在时,其据信为其中来自mgcl2的单个cl配位至al的物质(该配合物据信类似于在chien(1982)参考文献中归属于物质f的配合物);而当物质c存在并改为具有1.969的g值时,其据信为其中来自mgcl2的单个cl配位至ti的物质(该配合物据信类似于在chien(1982)参考文献中归属于物质e的配合物)。在一个实施方案中,本文中描述的主催化剂的固体组分包含至少0.2%、或例如约0.2%至约1%的具有1.950的g值的物质c,就在图4和5中所说明的意义而言。据信,该物质c在ti处具有四面体构型。在其他实施方案中,主催化剂具有约0.2%至约0.5%的具有1.950的g值的物质c,或者具有约0.5%至约1%的具有1.950的g值的物质c。相比之下,对比实施例a的固体组分使用其他的已知制备方法和在美国专利第7,666,810b2号中描述的方法来获得。对比实施例a的epr分析和模拟显示出存在具有1.969的g值的物质c(示于图6中),其可以归属为在ti处的三角双锥构型。不希望被束缚于任何特定的解释理论,已发现,当物质c以1.969的g值存在时,主催化剂与物质c以1.950的g值存在时的主催化剂相比,展现出在烯烃聚合活性方面和在高温聚合方法中生产的聚合物的分子量方面的更不利的特性。在一个实施方案中,主催化剂为式ticl3*[oetalcl2]d的ti3+配合物,并且mg/ti摩尔比为约5至约8。在另一个实施方案中,主催化剂为式ticl3*[clalcl2]d的ti3+配合物,并且mg/ti比为约5至约8。在一些实施方案中,可以存在ticl3*[clalcl2]d或ticl3*[oetalcl2]d的部分烷基化形式。在本文中描述的本发明的另一个实施方案提供用来制备包含ti3+配合物的烯烃聚合主催化剂的方法,所述方法包括:a)通过组合以下物质来形成δ型mgcl2物质:i)选自c5-12烷烃的溶剂中的r2mg,和ii)反应性有机氯化物或hcl;其中,每个r独立地选自c2-8烷基;并且其中所添加的cl与mg的摩尔比为约2.0至约3.0;然后,可替代地b)向步骤a)中制备的所述δ型mgcl2物质,以任意顺序或同时添加式r1xalx3-x的烷基铝卤化物和四价钛化合物,从而提供约3至约10的al/ti摩尔比;或者c)向步骤a)中制备的所述δ型mgcl2物质,首先添加式r1xalx3-x的烷基铝卤化物,并其次添加四价钛化合物,然后在最终添加步骤中添加式r4yalor53-y的烷基铝烷氧化物,其中,当测量由r1xalx3-x供应的al时的al/ti摩尔比为约0.7至约2,并且当测量由r4yalor53-y供应的al时的al/ti摩尔比为约1至约2;并且进一步地其中,mg/ti摩尔比为约5至约10,x为1或2,y为1或2,每个r1独立地选自c1-8烷基,四价钛化合物选自tir2x3、ti(or3)x3、tix4、和它们的混合物,每个x独立地选自卤素,每个r2独立地选自c1-8烷基和苄基,并且每个r3、r4和r5独立地选自c1-8烷基。在本文中描述的本发明的另一个实施方案提供通过以下方法制备的主催化剂产物,所述方法包括:a)通过组合以下物质来形成δ型mgcl2物质:i)选自c5-12烷烃的溶剂中的r2mg,和ii)反应性有机氯化物(rcl)或hcl;其中,每个r独立地选自c2-8烷基;并且其中所添加的cl与mg的摩尔比为约2.0至约3.0;然后,可替代地b)向步骤a)中制备的所述δ型mgcl2物质,以任意顺序或同时添加式r1xalx3-x的烷基铝卤化物和四价钛化合物,其中,al/ti摩尔比为约3至约10;或者c)向步骤a)中制备的所述δ型mgcl2物质,首先添加式r1xalx3-x的烷基铝卤化物,并其次添加四价钛化合物,然后在最终添加步骤中添加式r4yalor53-y的烷基铝烷氧化物,其中,当测量由r1xalx3-x供应的al时的al/ti摩尔比为约0.7至约2,并且当测量由r4yalor53-y供应的al时的al/ti摩尔比为约1至约2;并且进一步地其中,mg/ti摩尔比为约5至约10,x为1或2,y为1或2,每个r1独立地选自c1-8烷基,四价钛化合物选自tir2x3、ti(or3)x3、tix4、和它们的混合物,每个x独立地选自卤素,每个r2独立地选自c1-8烷基和苄基,并且每个r3、r4和r5独立地选自c1-8烷基。二有机镁二有机镁化合物是公知且商业上可获得的。二有机镁化合物通常可以由式mgr2表示,其中,每个r选自c2-8烃基。在一个实施方案中,每个r独立地选自直链c2-8烷基,包括但不限于乙基、丁基、己基和辛基。在另一个实施方案中,每个r独立地选自c2-4烷基。在另一个实施方案中,每个r独立地选自乙基和丁基。在一个实施方案中,mgr2选自丁基乙基镁(bem)、二丁基镁、和丁基辛基镁(bom)。在另一个实施方案中,mgr2是丁基乙基镁(bem)。二有机镁溶液是由albemarle销售的商业上可获得的材料。其他二有机镁化合物包括丁基乙基镁或二丁基镁的烃溶液(其可任选用有机铝化合物处理以改进溶解性和/或降低溶液粘度)。在一个实施方案中,mgr2提供于选自c5-12烷烃的溶剂中。在一个实施方案中,溶剂选自己烷、环己烷、癸烷、庚烷、异己烷、和十二烷、以及它们的混合物。在一个实施方案中,溶剂为异己烷。在一个实施方案中,溶剂为癸烷。在一个实施方案中,溶剂为庚烷。氯的量和氯源是在“镁-钛”聚合催化剂中使用二氯化镁是公知的。mgcl2通常被当做钛物质的载体。二有机镁化合物与2摩尔当量的氯反应以制备二氯化镁是用来制备催化剂载体的公知方法。本发明的实施方案使用二氯化镁载体,其通过二有机镁化合物(上文所述)与2至3摩尔当量的氯的反应来制备。在一个实施方案中,载体中的氯/镁比率为每摩尔镁约2.15至约3.0(基于起始二有机镁化合物中的镁的量)、或者约2.15至约2.5。氯源基本上自发地与二有机镁反应,并且是反应性的有机氯化物或hcl。在一个实施方案中,反应性的有机氯化物为c4-10叔烷基氯化物。在一个实施方案中,反应性的有机氯化物为叔丁基氯。在一个实施方案中,氯源为hcl。反应温度可为约20℃至约160℃、或者约40℃至约100℃、或者约50℃至90℃、或者约40℃至约90℃。按本文中公开来制备的mgcl2物质呈δ型,如通过测量x射线衍射测量的峰的半高度而确定。本领域技术人员已知δ型是α和β型mgcl2的高度无序的混合物。xrd谱在确定mgcl2载体的结构方面特别有用,所述结构通过通常以旋转变换无序(rototranslationaldisorder)为特征的结构的x射线谱来表征(参见例如g.natta等人,j.polym.sci.1961,51,399-410)。图1示出典型的来自α型的mgcl2的xrd谱。图2示出使用在本文中公开并要求保护的方法形成的δ型mgcl2的xrd图谱。在本文中描述的本发明的一些实施方案中,用于制备mgcl2物质的方法的优点使得主催化剂形成的下一步骤能够继续进行而不需要介入洗涤步骤(如果需要该步骤)。残留的二有机镁起始材料的有害影响通过以下方式而最小化:使起始材料反应从而达到所公开的cl与mg的摩尔比,或者用额外的氯源、例如异丁基alcl2来处理mgcl2。钛iv源然后,通过在上文描述的氯化镁载体上沉积钛化合物来制备本文中描述的主催化剂。起始钛(iv)化合物可以选自式tir2x3、ti(or3)x3、tix4的化合物,和它们的混合物,其中,每个r2选自c1-8烷基和苄基,并且r3选自c1-8烷基,并且每个x独立地为卤素。在一些实施方案中,卤素选自氯和溴。在其他实施方案中,卤素是氯。在一些实施方案中,r3选自c1-4烷基。在其他实施方案中,r3选自乙基、异丙基和叔丁基。在一些实施方案中,r2选自c1-4烷基。在其他实施方案中,r2选自乙基和异丁基。在一些实施方案中,r2为苄基。在一些实施方案中,四价钛化合物为ti(och2ch3)cl3、或ti(ch2ch3)cl3。在一些实施方案中,四价钛化合物选自ticl2br2和ticl4。在一些实施方案中,四价钛化合物为ticl4。将要被本领域技术人员理解的是,tir2x3、ti(or3)x3、tix4物质可以购买,或者替代地可以通过用公知的商业上可获得且廉价的烷基钛和烷氧基钛化合物反应来制备,所述烷基钛和烷氧基钛化合物例如为ti(r2)2x2、ti(r2)3x1、ti(or3)2x2、或ti(or3)3x1,其中,各个x、r2和r3如上文中所述。铝物质在本文中描述的方法中所使用的铝化合物在商业上购买于公司例如albemarle、sigma-aldrich、或fisherchemical。r1xalx3-x用于卤化二烷基镁化合物和格式试剂,并且以在上文中指定的摩尔比量进行添加从而使溶液中的过量卤素最小化并且使ti物质的过度还原最小化。在一些实施方案中,x为1。在其他实施方案中,x为2。在一些实施方案中,每个r1独立地选自甲基、乙基、丙基、异丙基、丁基和异丁基。在其他实施方案中,每个r1独立地为乙基和异丁基。尽管x可以为任意卤素,在一些实施方案中,x为cl或br。在其他实施方案中,x为cl。在本文中描述的制造主催化剂的方法的一个实施方案中,r1xalx3-x选自异丁基二氯化铝(ibadc)和乙基二氯化铝(eadc)。r4yalor53-y用于将钛物质还原成所需氧化态和/或可以与过量的卤化物反应。除此之外,该化合物可以充当用于在本文下文中公开的聚合反应的活化剂。除了r4yalor53-y物质之外,还可以使用上文描述的r1xalx3-x作为还原剂。其他还原剂包括alr*3、alr*2x、至alr*1x2,其中,r*为c2-8烷基。尽管r*可以为更高级的烷基,但这样的铝物质在商业上不合需要。在本文中描述的制造主催化剂的方法的一些实施方案中,r1xalx3-x为三异丁基铝。在一些实施方案中,y为2。在一些实施方案中,y为1。在一些实施方案中,每个r4和r5独立地选自c1-4烷基。在其他实施方案中,每个r4和r5为乙基。在本文中描述的制造主催化剂的方法的一个实施方案中,r4yalor53-y为二乙基乙醇铝(deal-e)。通过后续向mgcl2物质添加铝和钛物质来进行的主催化剂的制备可以通过替代的路径实现。在一个实施方案中,使用以任意顺序添加至钛化合物或者与钛化合物一起添加的r1xalx3-x化合物实现钛物质从ti4+至ti3+的还原。在该路径的一些实施方案中,al/ti摩尔比为约4至7。在该路径的其他实施方案中,al/ti比为约5。在另一个替代的路径中,在添加较少量的r1xalx3-x化合物(与在先前讨论的路径中使用的r1xalx3-x化合物的量相比)后,添加钛物质。还原为ti3+物质通过添加r4yalor53-y化合物来完成。在该路径的一些实施方案中,当测量由r1xalx3-x供应的al时的al/ti摩尔比为约1至约1.8。在该路径的其他实施方案中,当测量由r1xalx3-x供应的al时的al/ti摩尔比为约1。在该路径的一些实施方案中,当测量由r4yalor53-y供应的al时的al/ti摩尔比为约0.7至约1.7、或者约1.5至1.7。在该路径的其他实施方案中,当测量由r4yalor53-y供应的al时的al/ti摩尔比为约1.67。在所讨论的任一路径中,反应可以在约40℃至90℃、或者约40℃至约70℃、或者约45℃至约55℃的温度下进行,或者在约50℃的温度下进行。电子供体使用电子供体在基于镁-钛的烯烃聚合催化剂的领域中是公知的。本发明涵盖对电子供体的任选使用。但是,当在溶液聚合条件下使用催化剂时,优选不使用电子供体。适合的电子供体对本领域技术人员而言是公知的,并且包括四氢呋喃(thf)、二甲基甲酰胺、乙酸乙酯、甲基异丁酮和各种邻苯二甲酸酯/盐。活化剂可以在本发明中使用任何的“活化剂”,其活化上文描述的用于烯烃聚合的镁/钛主催化剂。示例性的活化剂包括铝氧烷和有机铝助催化剂。铝氧烷可以具有下式:(r6)2alo(r6alo)mal(r6)2其中,每个r6独立地选自c1-20烃基,并且m为0至50,优选r6为c1-4烷基,并且m为5至30。甲基铝氧烷(或“mao”)是优选的铝氧烷,其中每个r6为甲基。铝氧烷作为助催化剂,特别是用于茂金属型催化剂的助催化剂是公知的。铝氧烷也是易于获得的商业制品。使用铝氧烷助催化剂通常需要催化剂中的铝与过渡金属的摩尔比为25:1至1000:1。在本文中公开的方法中可用的示例比率为5:1至10:1。优选的有机铝化合物包括三乙基铝、三异丁基铝和二乙基乙醇铝。当使用这些有机铝活化剂时,基于主催化剂中的ti的摩尔数而言,示例性的al/ti比为0.5/1至10/1。溶液聚合法优选以相对低的al/ti摩尔比(例如0.5/1至5/1,特别是1/1至3/1)来进行,而气相聚合优选以相对高的al/ti摩尔比(例如20/1至150/1)来进行。用于乙烯的聚合和共聚的溶液法是本领域公知的。这些方法在惰性烃溶剂的存在下进行,所述惰性烃溶剂通常为c5-12烃,其可以未被取代或者被c1-4烷基取代,其例如为戊烷、甲基戊烷、己烷、庚烷、辛烷、环己烷、甲基环己烷和氢化石脑油。可商业上获得的合适溶剂的一个实例是“isopare”(c8-12脂族溶剂,exxonchemicalco.)。常规淤浆法或溶液法中的聚合温度为约80至约300℃(优选对于淤浆聚合而言为约80至约120℃,并且对于溶液聚合而言为约120至约250℃)。但是,如在实施例中例示的,用于本文中公开的溶液法的聚合温度可以高于160℃。温度上限将受到对本领域技术人员而言公知的考量的影响,例如期望使操作温度最大化从而降低溶液粘度,同时仍保持良好的聚合物特性。提高的聚合温度通常降低聚合物的分子量。在其他实施方案中,聚合温度可以为约200至约300℃,或者约220至约250℃。反应工艺的一个实例是“中压工艺”,其表示反应器中的压力优选小于约6,000psi(约42,000千帕斯卡或kpa)。压力可以为约10,000至约40,000kpa、或者约2,000至约3,000psi(约14,000-约22,000kpa)、或者725至约3,000psi(约5,000-约22,000kpa)。用于与乙烯共聚的合适单体包括c3-20单烯烃和二烯烃。示例共聚单体包括未被取代或者被最多两个c1-6烷基取代的c3-12α-烯烃、未被取代或被最多两个选自c1-4烷基的取代基取代的c8-12乙烯基芳族单体、未被取代或者被c1-4烷基取代的c4-12直链或环状二烯烃。这样的α-烯烃的说明性而非限制性的实例为以下中的一种或多种:丙烯、1-丁烯、1-戊烯、1-己烯、1-辛烯和1-癸烯、苯乙烯、α-甲基苯乙烯、和受限环(constrained-ring)环状烯烃,例如环丁烯、环戊烯、二环戊二烯、降冰片烯、烷基取代的降冰片烯、烯基取代的降冰片烯等(例如5-亚甲基-2-降冰片烯和5-乙叉基-2-降冰片烯、双环-(2,2,1)-庚-2,5-二烯)。乙烯与一种或多种可共聚单体的共聚物或三元共聚物还可以使用在本文中描述的方法来制备。在一个实施方案中,这样的聚合物将含有约50至约75重量%的乙烯、优选约50至60重量%的乙烯、以及相应的50至40重量%的丙烯。一部分单体、通常为丙烯单体可以被共轭二烯烃替代。二烯烃可以多至聚合物的10重量%的量存在,尽管通常以约3至5重量%的量存在。所得聚合物可以具有包含40至75重量%的乙烯、50至15重量%的丙烯、和多至10重量%的二烯单体的组成以提供100重量%的聚合物。二烯的优选但非限制性的实例是二环戊二烯、1,4-己二烯、5-亚甲基-2-降冰片烯、5-乙叉基-2-降冰片烯、和5-乙烯基-2-降冰片烯,特别是5-乙叉基-2-降冰片烯和1,4-己二烯。在另一个实施方案中,所得聚合物可以包含不小于约80重量%、或不小于约90重量%的乙烯,和最多约20重量%、或小于10重量%的一种或多种可共聚单体。在一些实施方案中,共聚单体为c3-8α-烯烃,例如1-丁烯、1-己烯和1-辛烯。单体在被进料至反应器之前溶解/分散在溶剂中(或者对于气态单体,可将单体进料至反应器使得其将溶解在反应混合物中)。在混合之前,可以对溶剂和单体进行纯化从而除去潜在的催化剂毒性物,例如水、氧气和其他极性杂质。原料纯化遵照本领域中的标准实践,例如使用分子筛、氧化铝床和除氧催化剂以纯化单体。优选还对溶剂本身(例如甲基戊烷、环己烷、己烷或甲苯)以类似方式进行处理。可以将原料在进料至反应器之前加热或冷却。在一些实施方案中,可以将催化剂组分预混合在用于反应的溶剂中,或者将其作为单独的物流进料至反应器。在一些情况中,将其预混合可能是合乎需要的从而在进入反应之前为催化剂组分提供反应时间。本文中描述的本发明的一个实施方案提供溶液烯烃聚合方法,其包括:i)使用上文中描述的方法来制备主催化剂;ii)将主催化剂与选自c5-12烷烃的溶剂添加至呈串联或并联配置的一个或多个反应器,连同向反应器添加乙烯和任选的选自c3-8共聚单体的一种或多种共聚单体、氢气;和iii)以相对于主催化剂的量为约1至约10的摩尔比将烷基铝活化剂添加至反应器。聚合方法还可以使用选自r4yalor53-y、三烷基铝化合物和mao的烷基铝活化剂。在一些实施方案中,在聚合方法中使用的溶剂选自己烷、环己烷、癸烷、庚烷、异己烷和十二烷。在其他实施方案中,溶剂为异己烷。在其他实施方案中,溶剂为癸烷。在一些实施方案中,溶液法在单个连续搅拌釜式反应器(cstr)中并任选以一个或多个附加的反应器来进行。在其他实施方案中,溶液法在以串联或并联形式安装的双反应器式连续反应器中进行。本发明的方法还可以包括使用与至少一个cstr的排出物相连的管式反应器。(为清楚起见,如果串联使用两个cstr,则管式反应器接收来自第二cstr的排出物)。术语“管式反应器”意在表达其常规含义——即简单的管。管式反应器可以具有至少10/1的长度/直径(l/d)比。管式反应器不受搅动,并且以绝热形式操作。因此,随着聚合进行,剩余的共聚单体越来越被消耗,并且溶液的温度升高(这两者改进了将剩余的共聚单体与聚合物溶液分离的效率)。沿着管式反应器的长度的温度增加可以大于3℃(即,来自管式反应器的排出物温度比来自对管式反应器进料的cstr的排出物温度大至少3℃)。管式反应器可以具有用于额外的乙烯和溶剂的进料口。进料为“经调温的”——即额外的乙烯和/或溶剂的温度被加热至高于环境(或者至约100℃),但所述温度低于管式反应器的排出物温度。在一个实施方案中,乙烯被调温至约80℃至约200℃、或者约100℃至约200℃。在一个实施方案中,与溶剂一起添加乙烯。溶剂的量(表述为基于乙烯的重量比)为约20/1至约0.1/1、或约10/1至约1/1。任选地,管式反应器还可以具有用于额外的催化剂、助催化剂、共聚单体和/或调聚剂(telomerizationagent)(例如氢气)的进料口。但是,在一些实施方案中,不向管式反应器添加额外的催化剂。管式反应器的总体积可以为所述至少一个cstr的体积的至少10体积%,或者约30%至约200%(为清楚起见,如果cstr的体积为约1000升,则管式反应器的体积为至少约100升,或约300至约2000升)。添加至管式反应器的乙烯的总量可以为添加至一个或多个cstr的总乙烯的1至50重量%。例如,如果以约1000kg/hr的乙烯流量操作一个cstr,则至管式反应器的乙烯流将为约10至约500kg/hr。类似地,如果以约1000kg/hr的至第一cstr的乙烯流量,和约500kg/hr的至第二cstr的乙烯流量操作两个cstr,则至管式反应器的乙烯流将为约15至约750kg/hr。在一些实施方案中,主催化剂是预配制的并且直接添加至反应器。在一些实施方案中,聚合温度为至少约220℃、或至少约230℃、或至少约240℃。在一些实施方案中,与使用专利美国专利第5589555号中公开的主催化剂的聚合方法相比,使用本文中描述的主催化剂的聚合方法产生具有相同密度的聚合物,但其中所述方法使用少至少约10%的共聚单体进料。在一些实施方案中,与使用不包含至少0.2%的具有1.950的g值的epr活性物质的主催化剂的聚合方法相比,使用本文中描述的主催化剂的聚合方法产生具有相同密度的聚合物,但其中所述方法使用少至少约10%的共聚单体进料。在一些实施方案中,与使用不含四面体ti3+物质、或者基本不含四面体ti3+物质的用于聚合的主催化剂的聚合方法相比,使用本文中描述的主催化剂的聚合方法产生具有相同密度的聚合物,但其中所述方法使用少至少约10%的共聚单体进料。基本上不含四面体ti3+物质意指存在少于约0.005%、或少于0.01%、或少于0.05%的四面体ti3+物质,如通过如本文中描述的epr和epr模拟所确定的。在其他实施方案中,针对使用美国专利第5589555号中公开的主催化剂的制备的聚合物,使用本文中描述的主催化剂的聚合方法产生具有相同密度,但在任意聚合温度下具有比前者聚合物所获得的mw更高的mw的聚合物。在其他实施方案中,针对使用不包含至少0.2%的具有1.950的g值的epr活性物质的主催化剂制备的聚合物,使用本文中描述的主催化剂的聚合方法产生具有相同密度,但在任意聚合温度下具有比前者聚合物所获得的mw更高的mw的聚合物。在其他实施方案中,针对使用不含四面体ti3+物质、或者基本不含四面体ti3+物质的用于聚合的主催化剂制备的聚合物,使用本文中描述的主催化剂的聚合方法产生具有相同密度,但在任意聚合温度下具有比前者聚合物所获得的mw更高的mw的聚合物。基本上不含四面体ti3+物质意指存在少于约0.005%、或少于0.01%、或少于0.05%的ti3+物质,如通过如本文中描述的epr和epr模拟所确定的。在一些实施方案中,反应器保持时间(hold-uptime)为约30秒至约1小时。在其他实施方案中,反应器保持时间为约30秒至约30分钟。在其他实施方案中,反应器保持时间为约30秒至约5分钟。在其他实施方案中,反应器保持时间为约1分钟至约5分钟。本发明的另一个实施方案提供具有约0.910g/cc至约0.935g/cc的密度的聚乙烯聚合物或共聚物。本发明的另一个实施方案提供大于或等于约50的cdbi50辛烯。本发明的另一个实施方案提供具有约3至约8的mwd的聚合物。本发明的又一个实施方案在最终聚合物产物中提供基本上平坦(flat)的共聚单体分布。基本上平坦的共聚单体分布意指如在gpc曲线上描绘的作为分子量的函数的支链含量描点将给出偏离水平不大于约15°的线。在一些实施方案中,聚合物在所得聚合物中具有少于约10ppm的计算残留钛。在其他实施方案中,聚合物在所得聚合物中具有少于约8ppm的计算残留钛。在其他实施方案中,聚合物在所得聚合物中具有少于约3ppm的计算残留钛。在一些实施方案中,聚合物在所得聚合物中具有少于约120ppm的计算残留卤素。在其他实施方案中,聚合物在所得聚合物中具有少于约100ppm的计算残留卤素。在其他实施方案中,聚合物在所得聚合物中具有少于约60ppm的计算残留卤素。本发明的另一个实施方案提供如在上文中描述的聚合物,其用于选自挤出、注射成型、热成型、和旋转成型的制造方法。本发明的另一个实施方案提供如在上文中描述的聚合物,其用于塑料制品,例如膜、纤维,模塑或热成型的制品,例如鼓状物和农业喷雾罐,以及管道涂层。将进一步通过参照以下实施例描述本发明。以下实施例仅是对本发明的例示,并且不意图进行限制。除非另有指明,所有百分比以重量计。实施例化学品和试剂将购买的环己烷通过使其经过脱氧催化剂床(品牌名r311,来自basf)、氧化铝床(品牌名selexsorbcos/cd)、和分子筛(3a/13x)床开进行干燥和脱氧。正癸烷购买自sigmaaldrich,并将溶剂转移至容纳有经活化的13x分子筛的nalgene瓶中,并在使用之前储存至少过夜。甲基戊烷购买自imperialoil,并且其包含100%的石油脑(石油)、经加氢处理的轻馏分(hydrotreatedlight)。通过使溶剂经过含有selexsorbcd和selexsorbcdx的床来将其干燥。丁基乙基镁(bem)(20wt.%在庚烷中的溶液)购买自albemarle。其容纳于pyrosafe筒中,并且储存在手套箱内。异丁基二氯化铝(ibadc)(97wt.%)购买自albemarle。其容纳于pyrosafe筒中,并且储存在手套箱内。ibadc具有242℃的沸点和1.12g/ml的密度。二乙基乙醇铝(deao)(25wt.%在庚烷中的溶液)购买自akzonobel。deao具有98℃的沸点和0.684g/ml的密度。乙基二氯化铝(eadc)(20wt.%在庚烷中)购买自akzonobel。eadc具有115℃的沸点和1.20g/ml的密度。二乙基氯化铝(deac)(97wt.%)购买自sigmaaldrich。deac具有125℃的沸点和0.961g/ml的密度。异丁基铝氧烷(ibao)(2.7wt.%在庚烷中)购买自albemarle。其容纳于玻璃瓶中,并且储存在手套箱冷冻器内。ibao具有98℃的沸点和0.691g/ml的密度。三异丁基铝(tibal)购买自akzonobel。tibal具有86℃的沸点和0.786g/ml的密度。干燥剂(drieritetm)购买自sigmaaldrich。在使用之前,通过将干燥剂在设置在260℃的马弗炉中烘烤16小时的时间来对其进行调节。干燥剂不含指示剂。2-氯-2-甲基丙烷(叔丁基氯或tbucl)购买自sigmaaldrich。通过在惰性环境下,以每100ml的tbucl30g干燥剂的比率,将tbucl放置在经预干燥的干燥剂上约16小时,从而干燥tbucl。在该处理过程中,将容纳tbucl的烧瓶用箔遮盖从而将其与光隔离,从而使异丁烯的形成最小化。通过真空转移来将经干燥的tbucl进一步纯化。纯化后,tbucl的水分含量为12ppm或更低,并具有大于97%的纯度。在该流程中使用的全部玻璃器具在130℃的烘箱中干燥过夜。乙烯按聚合级购买自praxair。通过使气体经过一系列纯化床来将乙烯纯化和干燥,所述纯化床包括氧化铝(品牌:selexsorbcos)、分子筛(类型:13x)和脱氧床(品牌:oxiclear®)。使购买的1-辛烯通过以1升批次储存在3a分子筛上来干燥。氯化钛(iv)(ticl4)在氮气下包装按99.9%纯度购买自sigmaaldrich。甲醇按gracs等级购买自emdchemicals。分析方法熔融指数(“mi”)测量根据astm方法d-1238来进行。聚合物密度使用astmd-1928来测量。聚合物分子量和分子量分布通过凝胶渗透色谱(gpc)来测量。仪器(waters150-c)在140℃下、在1,2,4-三氯苯中使用,并且使用聚乙烯标准物将其校准。聚合物支化频率(polymerbranchfrequencies)通过ft-ir来确定。所使用的仪器为nicolet750magna-ir分光光度计。针对钛价态分布对一些催化剂样品进行分析。基于科学论文(j.c.w.chien等人,j.polym.sci.parta:polym.chem.1989,27,1499-1514)开发了用于钛价态分布的氧化还原滴定法,并且基于astm标准e878-01开发了用于钛含量分析的紫外(uv)法。电感耦合等离子体质谱(icp-ms)分析在agilent7700系列的仪器上完成。将样品用5%的硝酸溶液消化,并在高能氦模式中分析从而除去任何的光谱干扰。仪器使用经认证的标准物来校准。ti和mg标准物购买自spcsciences,并且cl购买自bdh。使用brukergeneralareadetectordiffractionsystem(gadds)来收集x射线衍射图谱。x射线使用设置在30kv和30ma下的cu管(波长为1.54184a)来生成。样品至检测器距离为5.0cm。检测器至样品的角度(2θ)为30°。对于数据收集,将粉末化的样品置于1.0mmid石英管中。对衍射图谱进行背景校正。gpc-ft-ir:通过在烘箱中在150℃下将聚合物在1,2,4-三氯苯(tcb)中加热并在转轮(wheel)上旋转4小时,从而制备聚合物样品溶液(2至4mg/ml)。向混合物添加抗氧化剂2,6-二叔丁基-4-甲基苯酚(bht),从而使聚合物稳定化以免于氧化降解。bht浓度为250ppm。在watersgpc150c色谱装置上,在140℃下对样品溶液进行色谱分析,所述色谱装置装配有4个shodex柱(ht803、ht804、ht805和ht806),以1.0ml/分钟的流量使用tcb作为流动相,其中ftir光谱仪和经由经加热的传递管线而通过与色谱装置耦合的单元(cell)的经加热ftir流作为检测系统。以250ppm的浓度向流动相添加bht,从而保护sec柱免于氧化降解。样品注射体积为300ml。将原始ftir光谱用opusftir软件进行处理,并且与opus关联的chemometricsoftware(pls技术)来即时计算聚合物浓度和甲基含量。然后,获得聚合物浓度和甲基含量,并且用cirrusgpc软件对其进行基线校正。用窄分布的聚苯乙烯标准物来校准sec柱。使用mark-houwink公式将聚苯乙烯分子量换算为聚乙烯分子量,如在astm标准测试方法d6474中所描述的。基于通过如在由p.j.deslaurierspolymer2002,43,159-170出版的著作中描述的pls技术来预测的聚合物浓度和甲基含量,计算共聚单体含量。tref:将聚合物样品(80至100mg)引入至polymerchar晶体-tref装置的反应器容器中。用35ml1,2,4-三氯苯(tcb)填充反应器容器,将其加热至所需的溶解温度(例如150℃)2小时。然后,将溶液(1.5ml)载入填充有不锈钢珠的tref柱中。在给定的稳定化温度(例如110oc)下使其能够平衡45分钟后,在温度从稳定化温度降至30℃(0.09℃/分钟)的情况下使聚合物溶液能够结晶。在30℃下平衡30分钟后,用tcb(0.75ml/分钟)、以30℃至稳定化温度的温度斜升(0.25℃/分钟)洗脱结晶的样品。在运转结束时在溶解温度下清洁tref柱30分钟。使用polymerchar软件、excel电子表格和自行开发的tref软件来处理数据。cdbi定义为其组成在中值共聚单体组成(mediancomonomercomposition)的50%内的聚合物的百分比。其由组成分布曲线和组成分布曲线的标准化累计积分来计算,如在美国专利第5376439号中所例示的。epr是能够检测化学样品中的未成对电子的磁共振技术。其通过观察在用通常在微波频率范围内的单色辐射辐照时未成对电子共振所处于的磁场而发生。共振场的精确值对于电子的化学环境敏感,并且由g值来指定。g使用下式计算(atkins,peterw.,physicalchemistry,5thed.,1994,freemanpress,newyork):其中,h为普朗克常数(6.63×10-34j·s),ν为微波频率(以hz计),µb为玻尔磁子(9.27×10-24j·t-1),并且b为磁场(以t计)。全部epr谱均在室温下记录于brukeremx10/12光谱仪上。场校准使用强间距(strongpitch)标准物来完成。将齐格勒纳塔催化剂进行干燥并填塞在4mm石英epr管中,并用环氧树脂密封从而保持管内的惰性氛围。epr模拟使用基于matlab的软件包easyspin来进行。模拟的原理描述于论文(stefanstoll,arthurschweiger,“easyspin,acomprehensivesoftwarepackageforspectralsimulationandanalysisinepr”,j.magn.reson.2006,178,42-55)中。催化剂合成和表征所有实验均在手套箱中、在氮气氛围下使用油浴或者加热罩作为热源来进行。所使用的所有玻璃器具在碱浴中清洁过夜,在酸浴中冲洗,用去离子水冲洗,然后放置在135℃的烘箱中过夜以干燥。实施例1:mgcl2合成(δ-mgcl2)和表征:在1000ml圆底烧瓶中,将16.910g(31.377mmol)的20.5wt.%bem添加至295ml癸烷。然后,在使用顶置式搅拌器以350rpm进行搅拌的同时,将溶液加热至45℃的内温(使用偶丝(thermowire)来监测)。将稀释于5ml的癸烷中的7.149g(77.228mmol)的冷tbucl经由注射器一次性添加至bem溶液。将溶液在50℃下搅拌30分钟。在形成mgcl2浆料后,将整个混合物转移至500mlpyrex中。基于icp结果,cl/mg为2.33(mol/mol)。对于gadds结果,参见图2。实施例2:催化剂1的合成:在1000ml圆底烧瓶中,将16.909g(30.304mmol)的19.8wt.%bem添加至295ml癸烷。然后,在使用顶置式搅拌器以350rpm进行搅拌的同时,将溶液加热至45℃的内温(使用偶丝来监测)。将稀释于5ml癸烷中的7.072g(76.396mmol)冷tbucl经由注射器一次性添加至bem溶液。将溶液在50℃下搅拌30分钟。在形成mgcl2后,在50℃下,使用注射剂将0.758g(3.996mmol)的ticl4添加至mgcl2。在添加ticl4之后,接着经由滴液漏斗,以~3滴/秒的速率将稀释于20ml癸烷中的6.229g(40.1871mmol)ibadc添加至反应。一旦完成添加冲洗漏斗后,将浆料缓慢加热至85℃的内温,并将其搅拌1h。然后,经玻璃料(frit)过滤催化剂,用20ml癸烷洗涤1次,并用20ml环己烷洗涤4次,然后转移至100ml玻璃pyrex瓶中,并在80ml的环己烷中再浆化。来自催化剂1的epr谱和通过模拟进行的光谱反卷积(spectraldeconvolution)实验条件:频率=9.389ghz,微波功率=12.7mw,时间常数=0.64ms,调制幅度=1g,8次42s的扫描的平均。模拟参数:a)g=[1.897、1.907、1.944],线宽=[90、170、60]高斯;b)g⊥=1.880,g||=1.945,线宽(⊥)=290高斯,线宽(||)=330高斯;c)g=1.950,线宽=50高斯;对信号强度的贡献:a=18.6%,b=80.7%,c=0.7%。对于epr谱和模拟,参见图4。实施例3:催化剂2a的合成:在1000ml圆底烧瓶中,将16.665g(30mmol)的19.9wt.%bem添加至295ml癸烷。然后,在使用顶置式搅拌器以350rpm进行搅拌的同时,将溶液加热至45℃的内温(使用偶丝来监测)。将稀释于5ml癸烷中的6.942g冷tbucl经由注射器一次性添加至bem溶液。将溶液在50℃下搅拌30分钟。在形成mgcl2后,在50℃下,使用漏斗一次性将稀释于5ml癸烷中的1.059g(6.8mmol)二乙基氯化铝(ibadc)添加至mgcl2,并将溶液搅拌10分钟。在50℃下使用注射器将用5ml癸烷稀释的0.758g(4.0mmol)ticl4一次性全部添加至mgcl2。添加ticl4之后,经由移液管将稀释于5ml癸烷中的3.430g(6.6mmol)的25wt.%二乙基乙醇铝添加至反应。将浆料加热至85℃的内温,并且一旦溶液达到该温度,就将溶液搅拌1h。然后,经玻璃料过滤催化剂,用20ml癸烷洗涤1次,并用20ml环己烷洗涤4次,并然后转移至用于储存的玻璃瓶中,并在80ml的环己烷中再浆化。实施例4:催化剂2b的合成:向3000ml圆底烧瓶,将107.792g(200mmol)的20.5wt.%bem添加至约520ml的来自冷冻器的冷癸烷。然后,在使用顶置式搅拌器以470rpm进行搅拌的同时,将溶液加热至20℃的内温(使用偶丝来监测)。将稀释于70ml癸烷中的42.579g(460mmol)tbucl经由滴液漏斗一次性添加至bem溶液。一旦完成添加冲洗漏斗后,在将溶液加热至50℃的同时,将溶液搅拌35分钟。在形成mgcl2后,在50℃下使用滴液漏斗将稀释于30ml癸烷中的7.029g(45.3mmol)ibadc一次性添加至mgcl2。一旦完成添加冲洗漏斗后,将溶液搅拌10分钟。在添加ibadc之后,经由滴液漏斗将稀释于30ml癸烷中的5.057g(26.7mmol)ticl4一次性添加至反应。一旦完成添加冲洗漏斗后,将浆料搅拌5分钟。经由滴液漏斗将稀释于60ml癸烷中的22.904g(44mmol)deao一次性添加至反应。一旦完成添加冲洗漏斗后,将溶液缓慢加热至85℃的内温,并将其搅拌1h。关闭加热,并将溶液冷却30分钟。然后,经玻璃料过滤催化剂,用130ml癸烷洗涤1次,并用130ml环己烷洗涤2次。将固体催化剂转移至用于储存的玻璃瓶中,并用约350ml的环己烷再浆化。来自催化剂2b的epr谱和通过模拟进行的光谱反卷积。实验条件:频率=9.391ghz,微波功率=12.7mw,时间常数=0.64ms,调制幅度=1g,8次42s的扫描的平均。模拟参数:a)g=[1.899、1.899、1.949],线宽s=[120、120、120]高斯;b)g⊥=1.887,g||=1.945,线宽(⊥)=340高斯,线宽(||)=460高斯;c)g=1.950,线宽=42高斯;对信号强度的贡献:a=22.3%,b=77.5%,c=0.2%。对于epr谱和模拟,参见图5。实施例5:催化剂2c的合成:使用催化剂2b的流程,但在多批次合并的情况下制造催化剂。实施例6:催化剂3的合成:在1000ml圆底烧瓶中,将16.167g(30mmol)的20.5wt%bem添加至约285ml癸烷。然后,在使用顶置式搅拌器以345rpm进行搅拌的同时,将溶液加热至45℃的内温(使用偶丝来监测)。将稀释于5ml癸烷中的6.387g(69mmol)tbucl经由滴液漏斗一次性添加至bem溶液。一旦完成添加冲洗漏斗后,在将浆料加热至50℃的同时,将溶液搅拌30分钟。在形成mgcl2后,在50℃下使用滴液漏斗将稀释于5ml癸烷中的4.231g(6.8mmol)的20.4wt.%etalcl2一次性添加至mgcl2。一旦完成添加冲洗漏斗后,将溶液搅拌10分钟。在添加etalcl2之后,经由滴液漏斗将稀释于5ml癸烷中的0.761g(4.0mmol)的ticl4一次性添加至反应。一旦完成添加冲洗漏斗后,将浆料搅拌5分钟。经由滴液漏斗将稀释于10ml癸烷中的3.434g(6.6mmol)deao一次性添加至反应。一旦完成添加冲洗漏斗后,将溶液缓慢加热至85℃的内温,并将其搅拌1h。关闭加热,并将溶液冷却30分钟。然后,经玻璃料过滤催化剂,用20ml癸烷洗涤1次,并用20ml环己烷洗涤2次。将固体催化剂转移至用于储存的玻璃瓶中,并用约80ml的环己烷再浆化。通过icp对催化剂的wt%ti进行分析,并确定为4.06。实施例7:催化剂4的合成:在1000ml圆底烧瓶中,将16.167g(30mmol)的20.5wt%bem添加至约285ml癸烷。然后,在使用顶置式搅拌器以350rpm进行搅拌的同时,将溶液加热至45℃的内温(使用偶丝来监测)。将稀释于5ml癸烷中的6.387g(69mmol)tbucl经由滴液漏斗一次性添加至bem溶液。一旦完成添加冲洗漏斗后,在将浆料加热至50℃的同时,将浆料搅拌30分钟。在形成mgcl2后,在50℃下使用滴液漏斗将稀释于5ml癸烷中的0.825g(6.8mmol)二乙基氯化铝(deac)一次性添加至mgcl2。一旦完成添加冲洗漏斗后,将浆料搅拌10分钟。在添加deac之后,经由滴液漏斗将稀释于5ml癸烷中的0.755g(4.0mmol)ticl4一次性添加至反应。一旦完成添加冲洗漏斗后,通过漏斗全部一次性添加用5ml癸烷稀释的1.215g(2.33mmol)的25wt%deao,并且将浆料加热至85℃的内温,并将溶液搅拌1h。关闭加热,并将溶液冷却30分钟。然后,经玻璃料过滤催化剂,用50ml癸烷洗涤1次,并用20ml环己烷洗涤2次。将固体催化剂转移至用于储存的玻璃瓶中,并用约80ml的环己烷再浆化。通过icp对催化剂的wt%ti进行分析,并确定为3.905。实施例8:催化剂5的合成:向1000ml圆底烧瓶,将16.167g(30mmol)的20.5wt.%bem添加至约285ml癸烷。然后,在使用顶置式搅拌器以350rpm进行搅拌的同时,将溶液加热至45℃的内温(使用偶丝来监测)。将稀释于5ml癸烷中的6.395g(69mmol)tbucl经由滴液漏斗一次性添加至bem溶液。一旦完成添加冲洗漏斗后,在将浆料加热至50℃的同时,将浆料搅拌30分钟。在形成mgcl2后,在50℃下使用滴液漏斗将稀释于5ml癸烷中的1.052g(6.8mmol)ibadc一次性添加至mgcl2。一旦完成添加冲洗漏斗后,将浆料搅拌10分钟。在添加ibadc之后,经由滴液漏斗将稀释于5ml癸烷中的0.761g(4.0mmol)ticl4一次性添加至反应。一旦完成添加冲洗漏斗后,将浆料加热至85℃的内温。经约20分钟逐滴缓慢添加用癸烷稀释的10.054g(2.10mmol)的2.7wt%ibao。然后,将溶液搅拌1h。关闭加热,并将浆料冷却30分钟。然后,经玻璃料过滤催化剂,用20ml癸烷洗涤1次,并用20ml环己烷洗涤2次。将固体催化剂转移至用于储存的玻璃瓶中,并用约80ml的环己烷再浆化。通过icp对催化剂的wt%ti进行分析,并确定为3.33。实施例9:催化剂6的合成:向1000ml圆底烧瓶,将16.172g(30mmol)的20.5wt.%bem添加至约285ml癸烷。然后,在使用顶置式搅拌器以350rpm进行搅拌的同时,将溶液加热至45℃的内温(使用偶丝来监测)。将稀释于5ml癸烷中的6.389g(69mmol)tbucl经由滴液漏斗一次性添加至bem溶液。一旦完成添加冲洗漏斗后,在将浆料加热至50℃的同时,将浆料搅拌30分钟。在形成mgcl2后,在50℃下使用滴液漏斗将稀释于5ml癸烷中的1.060g(6.8mmol)ibadc一次性添加至mgcl2。一旦完成添加冲洗漏斗后,将浆料搅拌10分钟。在添加ibadc之后,经由滴液漏斗将稀释于5ml癸烷中的0.760g(4.0mmol)ticl4一次性添加至反应。一旦完成添加冲洗漏斗后,将浆料加热至85℃的内温。经约60分钟,逐滴缓慢添加用癸烷稀释的3.189g(4.10mmol)的25.7wt%tibal。然后,将溶液搅拌1h。关闭加热,并将溶液冷却30分钟。然后,经玻璃料过滤催化剂,用20ml癸烷洗涤1次,并用20ml环己烷洗涤2次。将固体催化剂转移至用于储存的玻璃瓶中,并用约80ml的环己烷再浆化。对比实施例1:催化剂7的合成:在1000ml圆底烧瓶中,将16.665g(30mmol)的19.9wt.%bem添加至约285ml癸烷。然后,在使用顶置式搅拌器以350rpm进行搅拌的同时,将溶液加热至45℃的内温(使用偶丝来监测)。将稀释于5ml癸烷中的6.942g(75mmol)冷tbucl经由注射器一次性添加至bem溶液。在将浆料加热至50℃的同时,将浆料搅拌30分钟。在形成mgcl2后,在50℃下使用注射器将稀释于5ml癸烷中的0.758g的ticl4一次性全部添加至mgcl2。在添加ticl4之后,在将浆料加热至85℃的同时,经由移液管将稀释于5ml癸烷中的5.880g(11.3mmol)25wt.%二乙基乙醇铝添加至反应(每次1-2ml)。在每次添加之间,将浆料搅拌15分钟,并且温度缓慢升高至85℃(即,在50、60、70℃下添加直至颜色不再变化)。当颜色不再变化时,停止添加。一旦其达到温度,将浆料搅拌1h。然后,经玻璃料过滤催化剂,用20ml癸烷洗涤1次,并用20ml环己烷洗涤4次。然后,将固体催化剂转移至用于储存的玻璃瓶中,并用约80ml的环己烷再浆化。对比实施例2:催化剂8的合成:在母液去除而无洗涤步骤的情况下,完全按催化剂2制造催化剂。基于us7,666,810b2中的公开内容的对比实施例a在手套箱内,在1000ml圆底烧瓶中,将16.665g(30mmol)的19.9wt.%bem添加至100ml环己烷。然后,将溶液进一步用另外的190ml环己烷稀释,并置于油浴中。烧瓶装配有含热偶丝的冷凝器、浆式搅拌器和间隔物。使用顶置式搅拌器,将溶液以400rpm搅拌并加热至45℃。经由气密注射器,一次性添加稀释于约5ml环己烷中的5.95ml(54mmol)tbucl。将溶液在50℃下搅拌半小时。过滤催化剂浆料并用环己烷洗涤3次(每次20ml)。使用注射器,以mg:ti=7.5的比率,在50℃下将1.79ml的2.24mticl4溶液添加至astmgcl2。将溶液搅拌半小时,然后过滤并用环己烷洗涤3次(每次20ml)。将催化剂在70ml的环己烷中再浆化,并转移至用于储存的玻璃瓶中。将少量的催化剂浆料干燥,并制备固体样品用于epr分析。来自制备对比实施例a的epr谱和通过模拟进行的光谱反卷积:实验条件:频率=9.395ghz,微波功率=12.7mw,时间常数=0.64ms,调制幅度=1g,8次42s的扫描的平均。模拟参数:a)g=[1.910、1.898、1.955],线宽s=[175、115、75]高斯;b)g⊥=1.883,g||=1.972,线宽(⊥)=235高斯,线宽(||)=200高斯;c)g=1.969,线宽=37高斯;对信号强度的贡献:a=52.4%,b=47.3%,c=0.3%。对于epr谱和模拟,参见图6。表1催化剂特性在实验室规模的连续式聚合装置中测试离线(offline)催化剂连续式聚合在连续式聚合装置(cpu)上进行。cpu包含71.5ml搅拌反应器,并且对聚合实验而言,在160至280℃运行。具有20ml体积的上游混合反应器在比聚合反应器低5℃的温度下运行。使用混合反应器来预热乙烯、辛烯和一些溶剂物流。以连续过程直接向聚合反应器添加催化剂进料和剩余的溶剂。保持27ml/分钟的进入聚合反应器的总连续流量。在浆料递送系统中,将来自上述实施例的催化剂添加至cpu。浆料递送系统由带有3500ml搅拌浆料储存器的倒置1000ml注射泵构成。浆料通过压差从搅拌瓶经由不锈钢套管而转移至3500ml搅拌浆料储存器中。然后,在储存器中将浆料用经纯化的环己烷稀释至所需浓度。一旦将浆料转移并稀释,在将任何浆料转移至注射泵中之前,将其在储存器中搅拌最少15分钟。当浆料就绪以转移至反应器时,打开气动电磁阀以使得浆料流动至注射器筒,所述气动电磁阀将储存器与注射器筒相隔离。然后,在注射器筒中恒定搅拌的情况下,以25ml/分钟的流量将注射器筒装载至期望的体积。当将注射器筒填充至所需体积时,关闭至储存器的电磁阀,将注射器筒和电磁阀相隔离。然后,在仍与反应器相隔离的同时,使注射器筒达到反应器压力。当注射器筒已达到反应器压力时,打开气动电磁阀(其将注射器筒与反应器相隔离)。然后,校准注射泵,并进行程序控制从而递送所需的浆料流量。对于浆料催化剂实验,以0.5的辛烯/乙烯重量比来制造共聚物。在聚合反应器中,以10wt.%的乙烯浓度进料乙烯。cpu系统在10.5mpa的压力下运行。在进入反应器之前,溶剂、单体和共聚单体物流均通过cpu系统纯化。q是乙烯转化率(如通过在线气相色谱(gc)而确定),并且聚合活性kp定义如下:(kp)(hut)=q((1-q)(1/催化剂浓度)其中,q为所转化的乙烯单体的分数;hut是聚合反应器中的倒易空间速度(保持时间),以分钟表达,并且贯穿整个实验程序保持恒定;并且催化剂浓度为聚合反应器中的浓度,以mmolti/l表达,并且浆料催化剂的ti浓度通过icp确定。所有聚合实验在220℃下进行,并且以90±1的乙烯转化率和2至4的二乙基乙醇铝(deao)与ti的摩尔比收集聚合物。表2cpu上的催化剂性能运行#催化剂代号cj/ti比率乙烯转化率qkp(1/mm*分钟)mw(10-3)pdbr/1000c1催化剂13.690.259.066.12.710.22催化剂2a2.089.789.857.43.012.43催化剂2b3.390.562.573.63.510.54催化剂2c2.289.771.768.73.011.15催化剂32.290.669.370.13.110.96催化剂42.190.674.966.62.610.07催化剂52.390.068.967.92.910.98催化剂62.089.668.878.42.710.79催化剂71.590.457.165.13.39.710催化剂82.189.972.961.83.111.7对以试验性工厂(pilotplant)规模的连续聚合设施进行的本发明的离线齐格勒纳塔(z/n)浆料催化剂(催化剂2c)和作为对比实施例3的通过管线内形成的zn催化剂制造的催化剂9的测试。表3中的实施例例示了使用单个试验性工厂反应器系统和离线浆料催化剂(催化剂2c)在中压下的乙烯和1-辛烯的连续流溶液共聚合。第一反应器为具有24.0升体积的连续搅拌釜式反应器(cstr)。第二反应器为具有cstr体积的10%的体积(2.4升)的管式反应器。如表3中说明地,向cstr中进料单体、溶剂和催化剂(操作模式1)。在实验中,使用具有由二乙基乙醇铝(deao)或三异丁基铝(tibal)构成的活化剂的离线齐格勒纳塔浆料催化剂(催化剂2c)。为了比较本发明的催化剂2c,使用对比的管线内形成的齐格勒纳塔(z/n)催化剂体系(催化剂9),并且在下一部分对其进行描述。使用浆料递送系统,将催化剂2c泵入连续流聚合反应器。浆料递送系统由浆料筒、搅动浆料日量槽、再循环回路、浆料催化剂计量泵、和溶剂稀释剂回路构成。通过用氮气对筒进行加压/鼓泡,以数次装料将经稀释的浆料催化剂从浆料筒转移至浆料日量槽。一旦将浆料催化剂转移至浆料催化剂日量槽中,启动搅动器和再循环泵以将催化剂浆料保持悬浮和恒定组成。将经稀释的浆料催化剂的温度保持在环境温度。将槽压力保持在300kpag。当浆料催化剂就绪以转移至反应器时,启动浆料催化剂递送泵,并且将浆料催化剂集中至泵。在浆料催化剂递送泵的排出口处,使用高流动溶剂(highflowsolvent)稀释剂从而使浆料催化剂保持悬浮,并且辅助催化剂向反应器的递送。将稀释剂流量保持在15kg/hr。将溶剂的温度控制在25℃。将溶剂和浆料催化剂泵送至流量传感器,并且记录流量。进入反应器的浆料催化剂流量通过稀释剂流量与合并的稀释剂和浆料催化剂的流量之间的差来计算。进入反应器的浆料催化剂流量(和ppms)通过改变浆料催化剂递送泵的马达变频驱动、或者泵推送器(pumpstroker)来调节。催化剂流量具有如在表中示出的表达为ti的按重量计的百万分率的目标设定值,并且对其进行调节以将总乙烯转化率保持为大于80%。使用如上提及的管线内形成的齐格勒纳塔催化剂体系(催化剂9),其由四氯化钛(ticl4)、丁基乙基镁(bem)和叔丁基氯(tbucl)构成,具有由三乙基铝(teal)或二乙基乙醇铝(deao)构成的活化剂。bem和teal以“预混合”(20/1mg/al摩尔比)的形式提供。在catalysttorpedo内,将所有催化剂组分混合于甲基戊烷溶剂中。混合顺序为bem/teal和tbucl(段#1);接着ticl4(段#2);然后接着deao(段#3)。将催化剂与甲基戊烷溶剂一同泵送至反应器中。催化剂流量具有表达为ti的按重量计的百万分率的目标设定值,并且对其进行调节以将总乙烯转化率保持为大于80%。因此,在表3中,对于给定的熔融指数、密度和应力指数,产物1、即在该反应器配置中制备的产物建立了“基线”反应器操作条件。产物2使用具有相同的铝活化剂的催化剂2c来制造。将反应器操作条件调节至生成与产物3中相似的熔融指数、密度和应力指数,产物3使用三异丁基铝(tibal)作为活化剂来制备。表4中的实施例例示了使用单个试验性工厂反应器系统和离线浆料催化剂(催化剂2c)在中压下的乙烯和1-辛烯的连续流溶液共聚合。第一反应器为具有24.0升体积的连续搅拌釜式反应器(cstr)。第二反应器为具有cstr体积的82%的体积(19.7升)的管式反应器。将催化剂进料至cstr中。如表4中说明地,将单体和溶剂在两个反应器之间拆分(操作模式2)。为了对比,使用管线内形成的齐格勒纳塔催化剂体系(催化剂9)。因此,在表4中,对于给定的熔融指数、密度和应力指数,产物4、即在该反应器配置中制备的产物建立了“基线”反应器操作条件。产物5使用具有相同的活化剂的离线浆料催化剂(催化剂2c)来制造。将反应器操作条件调节至生成与产物4中相似的熔融指数、密度和应力指数。产物6使用三异丁基铝(tibal)作为活化剂而生成。在表中使用的其他缩写的列表如下:hr:小时conc:浓度wt%:重量百分比wt/wt:重量/重量temp:温度c:摄氏度rpm:转/分钟mol:摩尔或摩尔的ppm:按重量计的百万分率。表3在操作模式1下的催化剂2c和催化剂9的性能表4在操作模式2下的催化剂2c和催化剂9的性能聚合物特性通过gpc-ft-ir和针对cdbi的tref进一步对产物5进行表征。gpc-ft-ir示出相对平坦的共聚物引入,并且来自催化剂2c的产物5的cdbi为58.4。由产物2、3、5和6以及来自对比实施例3和4的产物1和4来制备膜。在常规的吹塑生产线上制造所述膜,其通过具有2.5英寸螺杆直径的单螺杆挤出机来进料。通过电动马达来驱动挤出机。向全部挤出操作添加常规的添加剂(抗氧化剂和加工助剂)。迫使挤出物穿过具有4英寸直径和35mil模具间隙的圆形模具。使用2.5:1的吹胀比(bur)从而以100lbs/hr的输出率制备膜。对于1mil膜,产物2、3、5和6以及对比实施例3和4中的产物1和4的膜特性(例如落镖冲击、1%割线模量、md撕裂、td撕裂、雾度和己烷可提取性)基本上相同(在实验误差内)。工业实用性本发明涉及用于聚烯烃的高温溶液聚合的镁-钛催化剂。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 用于烯烃聚合的催化剂组分及其制备方法和应用和用于烯烃聚合的催化剂及其应用与制造工艺
- 用于烯烃聚合的催化剂组分及其制备方法和应用和用于烯烃聚合的催化剂及其应用与制造工艺
- 催化剂组分及其制备方法和应用和用于烯烃聚合的催化剂体系及其应用和烯烃聚合方法与制造工艺
- 用于烯烃聚合的催化剂组分及其制备方法和应用和用于烯烃聚合的催化剂及其应用与制造工艺
- 用于烯烃聚合的催化剂组分及其制备方法和应用和用于烯烃聚合的催化剂及其应用与制造工艺
- 用于烯烃聚合的催化剂组分及其制备方法和应用和用于烯烃聚合的催化剂及其应用与制造工艺
- 用于烯烃聚合的催化剂组分及其制备方法和应用和用于烯烃聚合的催化剂及其应用与制造工艺
- 催化剂组分及其制备方法和应用和用于烯烃聚合的催化剂体系及其应用和烯烃聚合方法与制造工艺
- 一种回收甲烷磺酸的装置的制造方法
- 一种用于生产高熔体流动速率聚烯烃的含杂环类化合物的烯烃聚合反应催化剂体系的制作方法