改进的核酸定量方法与流程
本发明涉及在不需要将数据标准化至感兴趣的参照基因或合成基因下定量核酸的方法。特别地,本发明涉及用于基因表达研究的改进的定量核酸的通用方法。该方法适用于诊断、法医和研究用途。然而,应当理解,本发明不限于这些特定领域的用途。
背景技术:
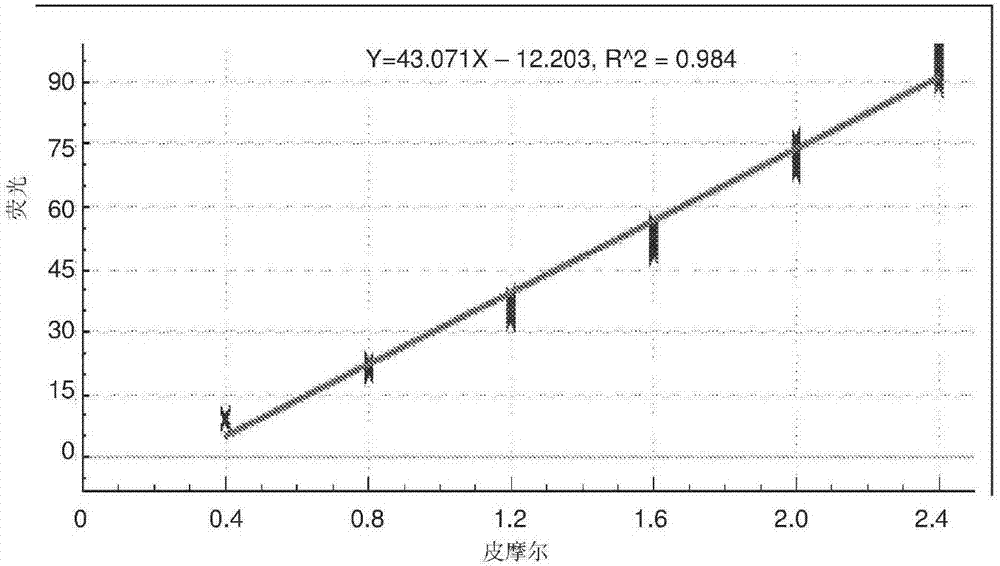
:在整个说明书中对现有技术的任何讨论绝不应被认为是承认这种现有技术是广为人知的或构成本领域公知常识的一部分。通过开发快速热循环仪以及在每次循环后引入扩增产物的荧光监测(实时pcr)而使得用于定量基因表达的pcr技术得以改善。通过使用染料,特别是荧光染料,以及在与反应开始时样品中核酸的量成比例的pcr扩增指数期检测荧光增加,而发生基因表达的定量。定量是基于阈值循环,即具有可检测荧光的第一次循环,并且可以以利用外部标准品(通常为合成基因)的绝对方式或以利用比较性的标准化参照基因作为内部校准物的相对方式(即,参照基因)进行定量。对照基因或参照基因用于将不同样品之间的mrna水平标准化。由于基因表达的变化会改变靶基因的相对定量分布,所以选择的参照基因不会波动是至关重要的。移液和稀释误差也会改变扩增水平,从而改变定量分布。经常使用如甘油醛-3-磷酸脱氢酶(gapdh)、胆色素原脱氨酶(pbgd)、β2-微球蛋白(b2m)或β-肌动蛋白(actb)的基因作为实时pcr中的内部校准物。然而,已经显示上述基因应答于实验条件或处理而改变表达水平。丰富表达的基因(如18s)也不是理想的参照基因,由于需要限制pcr条件,从而不会陷入(swamp)反应。因此,合适的参照基因应当在感兴趣的组织中充分表达,最重要的是,在样品之间以及在所使用的实验条件或处理下表现出最小的表达变异性。然而,这些对照基因中的许多可能表现出不可接受的表达变异性。已经表明,这些基因的表达水平可以在组织或细胞中变化,并且可能在某些情况,即在不同的处理条件下变化。因此,在任何新的实验系统中验证参照基因是至关重要的。找到适合在特定实验系统中使用的参照基因通常是耗时且困难的任务。在某些情况下,这可能是不可能的。在基因表达研究中使用外部标准品(即,合成参照)通常需要克隆感兴趣的基因以提供合成的参照基因。在该方法中,将已知量的合成参照基因序列系列稀释,然后进行扩增以产生标准曲线。该方法的克隆序列的生产通常是耗时、劳动密集的任务,并且稀释误差以指数方式放大,这可能导致不准确地评估核酸水平。高度稀释的克隆序列的稳定性和保存也会引起困难。因此,仍然需要定量核酸的简单而有效的通用方法,该方法适用于不需要使用感兴趣的参照基因或合成基因以标准化数据和/或定量基因表达的任何实验情况或处理条件。本发明的目的是克服或改善现有技术的至少一个缺点,或提供有用的替代方案。技术实现要素:在广泛的方面,本发明涉及用于定量感兴趣的核酸而不需要使用感兴趣的参照基因或合成基因来标准化核酸表达数据的方法。当然,本发明的方法利用可用于产生校准曲线的通用参照核酸,由该校准曲线可以计算样品中经过扩增的靶核酸的水平。本发明还涉及用于本发明方法的试剂盒。在第一方面,本发明提供了定量靶核酸的方法,该方法包括以下步骤:测量已知量的荧光团标记的通用参照核酸的荧光以产生校准曲线;在荧光团标记的探针的存在下扩增靶核酸,其中该荧光团标记的探针与靶核酸杂交;测量在靶核酸扩增期间的荧光;将扩增期间的荧光与校准曲线进行比较并确定经过扩增的靶核酸的量。在一个实施方式中,基本上同时进行步骤(a)至(d)。在另一个实施方式中,荧光团标记的通用参照核酸是单链。在另一个实施方式中,使用约0nm至约500nm的荧光团标记的通用参照核酸产生校准曲线。在另一个实施方式中,使用约0nm至约200nm的荧光团标记的通用参照核酸产生校准曲线。在另一个实施方式中,使用至少三种已知量的荧光团标记的通用参照核酸产生校准曲线。在另一个实施方式中,使用至少六种已知量的荧光团标记的通用参照核酸产生校准曲线。在另一个实施方式中,使用约0nm、约20nm、约40nm、约60nm、约80nm、约100nm和约120nm的荧光团标记的通用参照核酸产生校准曲线。在另一个实施方式中,使用约0nm、约20nm、约40nm、约60nm、约80nm、约100nm、约120nm、约140nm、约160nm和约200nm的荧光团标记的通用参照核酸产生校准曲线。在另一个实施方式中,通用参照核酸的长度小于约60bp。在另一个实施方式中,通用参照核酸的长度大于约170bp。在另一个实施方式中,通用参照核酸的长度约20bp。在另一个实施方式中,通用参照核酸的gc含量小于约45%、优选小于约40%、优选小于约35%、优选小于约30%、优选小于约25%、优选小于约20%、优选小于约15%、优选小于约10%。在另一个实施方式中,通用参照核酸的gc含量大于约60%、优选大于约65%、优选大于约70%、优选大于约75%、优选大于约80%、优选大于约85%、优选大于约90%。在另一个实施方式中,通用参照核酸的gc含量为约30%。在另一个实施方式中,通用参照核酸的gc含量为约40%。在另一个实施方式中,通用参照核酸的gc含量为约60%。在另一个实施方式中,通用参照核酸的gc含量为约80%。在另一个实施方式中,通用参照核酸的长度为约21bp以及gc含量为约62%。在另一个实施方式中,靶核酸的长度小于约80bp。在另一个实施方式中,靶核酸的长度大于约150bp。在另一个实施方式中,靶核酸的长度大于约210bp。在另一个实施方式中,靶核酸的长度约90bp。在另一个实施方式中,靶核酸的长度约500bp。在另一个实施方式中,靶核酸的长度约1000bp。在另一个实施方式中,靶核酸的gc含量约75%。在另一个实施方式中,靶核酸的gc含量约25%。在另一个实施方式中,靶核酸的扩增是通过聚合酶链式反应(pcr)进行的。在另一个实施方式中,pcr是实时pcr。在另一个实施方式中,实时pcr是多重pcr。在另一个实施方式中,荧光团标记的通用参照核酸和荧光团标记的探针在相同的发射通道中是可检测的。在另一个实施方式中,荧光团标记的通用参照核酸和荧光团标记的探针具有相同的激发和发射光谱。在另一个实施方式中,荧光团选自由fam、hex、德克萨斯红(texasred)、tye665、alexa594和cy5所组成的组。在另一个实施方式中,进行pcr约30至约45次循环。在第二方面,本发明提供了一种试剂盒,包括:(a)一种或多种的荧光团标记的通用参照核酸;(b)一种或多种的荧光团标记的探针;和(c)用于在第一方面的方法中使用的说明书。在第三方面,本发明提供了一种在第一方面的方法中使用时的试剂盒,该试剂盒包含:(a)一种或多种的荧光团标记的通用参照核酸;和(b)一种或多种的荧光团标记的探针。通用参照核酸序列不需要与靶核酸或与任何参照基因序列具有任何同源性。然而,由于在本发明的方法中使用通用参照核酸的特定方式(即,在将通用参照核酸系列稀释之后制备校准曲线),该通用参照核酸序列可以具有一定程度的同源性,或者甚至可以与靶核酸序列或参照基因或其较小部分相同。本发明的优点在于该方法可以利用相同的通用参照核酸来定量不同的靶核酸。通用参照核酸可以与靶核酸序列具有相同或相似的长度,但不一定如此。通用参照核酸可以比靶核酸更长或更短。通用参照核酸可以是双链或单链。使用已知技术可以从生物来源、天然的或其它来源获得通用参照核酸,或者其可以是合成制备的。通用参照核酸和探针均用荧光团标记。本领域技术人员已知的合适荧光团,包括但不限于fam、joe、hex、alexa594、德克萨斯红、cy5和tye665。当在实时pcr中使用时,应选择荧光团以使其在实时pcr装置的发射通道中是可检测的。优选地,使用通用参照核酸制备单个校准曲线,并且将校准曲线用于多种靶核酸扩增和定量。优选地,通过聚合酶链式反应(pcr)方法进行靶核酸的扩增。通常,将靶核酸扩增30至40次循环,但这对于本发明的方法并不是至关重要的。也可以在一次反应中同时扩增多种感兴趣的靶核酸(即,多重pcr)。除非上下文清楚地要求,否则在整个说明书和权利要求书中,词语“包含(comprise)”,“包括(comprising)”等将以包容性意义进行解释,与排外性或穷尽的意义相反;也就是说,在“包括但不限于”的意义上。附图说明图1:对于稀释至0nm、20nm、40nm、60nm、80nm、100nm和120nm的21bp和30%gc含量的fam标记的参照核酸,在40次pcr循环内测量的荧光(图1a)。通过绘制每次循环的荧光值相对于参照核酸浓度而创建校准曲线,并且计算线性回归(图1b)。使用fam标记的水解探针,在对来自系列稀释的原种λdna的92bp片段的扩增期间在40次pcr循环内测量的荧光(图1c)。图2:对于稀释至0nm、20nm、40nm、60nm、80nm、100nm和120nm的21bp和40%gc含量的fam标记的参照核酸,在40次pcr循环内测量的荧光(图2a)。通过绘制每次循环的荧光值相对于参照核酸浓度而创建校准曲线,并且计算线性回归(图2b)。使用fam标记的水解探针,在对来自系列稀释的原种λdna的92bp片段的扩增期间在40次pcr循环内测量的荧光(图2c)。图3:对于稀释至0nm、20nm、40nm、60nm、80nm、100nm、120nm、140nm、160nm和200nm的21bp和62%gc含量的fam标记的参照核酸,在40次pcr循环内测量的荧光(图3a)。通过绘制每次循环的荧光值相对于参照核酸浓度而创建校准曲线,并且计算线性回归(图3b)。使用fam标记的水解探针,在对来自系列稀释的原种λdna的92bp片段的扩增期间在40次pcr循环内测量的荧光(图3c)。图4:对于稀释至0nm、20nm、40nm、60nm、80nm、100nm和120nm的21bp和80%gc含量的fam标记的参照核酸,在40次pcr循环内测量的荧光(图4a)。通过绘制每次循环的荧光值相对于参照核酸浓度而创建校准曲线,并且计算线性回归(图4b)。使用fam标记的水解探针,在对来自系列稀释的原种λdna的92bp片段的扩增期间在40次pcr循环内测量的荧光(图4c)。图5:使用fam标记的水解探针(图5a)或嵌入染料evagreen(图5b),在对来自系列稀释的原种λdna的92bp片段的扩增期间的40次pcr循环内测量的荧光。图6:使用fam标记的水解探针,在对来自系列稀释的原种λdna的92bp片段(图6a)、501bp片段(图6b)或1000bp片段(图6c)的扩增期间在40次pcr循环内测量的荧光。图7:使用fam标记的水解探针,在对来自系列稀释的原种合成寡核苷酸的具有75%cg组成(图7a)或75%at组成(图7b)的90bp片段的扩增期间的40-45次pcr循环内测量的荧光。图8:对于系列稀释的fam(图8a)、hex(图8b)、德克萨斯红(图8c)和tye665(图8d)标记的参照核酸在45次pcr循环内测量的荧光。通过绘制每次循环的荧光值相对于荧光团标记的参照核酸浓度,创建针fam(图8e)、hex(图8f)、德克萨斯红(图8g)和tye665(图8h)的校准曲线。图9:使用fam标记的水解探针(图9a)、hex-(黑)或joe-(灰)标记的水解探针(图9b)、alexa594-(黑)或德克萨斯红-(灰)标记的水解探针(图9c)或者cy5-(黑)或tye665-(灰)标记的水解探针(图9d),在对稀释的λdna的扩增期间的45次pcr循环内测量的荧光。图10:采用单独使用(黑)或同时使用(灰)fam-、hex-或德克萨斯红-标记的水解探针,在对来自系列稀释的λdna的92bp片段的扩增期间的40次pcr循环内测量的并在绿色(fam)通道(图10a)、黄色(hex)通道(图10b)或橙色(德克萨斯红)通道(图10c)中检测的荧光。图11:使用fam标记的探针在对mirna的扩增期间的40次pcr循环内测量的荧光。具体实施方式在本发明的上下文中的核酸是由核苷酸链组成的分子。如本文中所使用的,该术语旨在包括dna、rna及其变体和衍生物。核酸可以是双链或单链。在本发明的上下文中的靶核酸是感兴趣的核酸。可以通过聚合酶链式反应(pcr)方便地完成核酸序列的扩增,但是也可以通过其它合适的方法如连接酶链式反应来完成。在本说明书的上下文中,根据本领域技术人员所理解的其常规含义使用术语“聚合酶链式反应”及其首字母缩写“pcr”。可以在本领域中使用的普通分子生物学教科书和参考手册中找到pcr方法的实例。例如,pcrtechnology:principlesandapplicationsfordnaamplification(1989)haerlich编辑.stockton出版,纽约。为了优化pcr扩增,可以以不同的浓度和比率使用引物。这些和其它变量的选择将在本领域技术人员的能力范围内得到理解。在本发明的上下文中的扩增子是通过扩增反应(如通过pcr或连接酶链式反应进行的那些)形成的核酸。在一个上下文中,扩增子可以是“靶核酸”的扩增产物。在本发明的上下文中的实时pcr是基于pcr的实验室技术,其用于同时扩增和检测/定量扩增子。在本发明的上下文中的多重pcr是基于pcr的实验室技术,其由在单个pcr混合物内的多种引物组和探针组成,以产生对不同靶核酸特异的多种扩增子。在本发明的上下文中的探针是可变长度的核酸,其用于检测与探针中的序列互补的靶核酸的存在。探针包括但不限于水解探针、分子信标和蝎型探针。在本发明的上下文中的引物是能够作为核苷酸合成的初始点的核酸。在本发明的上下文中的通用参照核酸包括可用于制备用于定量靶核酸的校准曲线的任何核酸,其中相同的通用参照核酸可用于定量不同的靶核酸。通用参照核酸可以是完全合成的或者可以从天然来源的核酸获得。在本发明的上下文中的荧光团是在光激发时可以重新发光的荧光化学分子。在本发明的上下文中的参照基因通常是维持基础细胞功能所需的组成型基因。所有细胞均存在这些基因。一些参照基因以相对恒定的水平表达,然而其它参照基因的表达可能根据所采用的实验条件而改变。在本发明的上下文中的校准曲线是荧光相对于荧光团标记的通用参照核酸的量的曲线图。在本发明的上下文中的标准曲线是循环阈值(ct或cq)相对于系列稀释的感兴趣的合成基因或输入核酸的曲线图。在本发明的上下文中的系列稀释是指制备包括感兴趣的荧光团标记的通用参照核酸或合成基因的一定浓度范围的校准曲线或标准曲线所必需的任何形式的稀释。在本发明的上下文中,“dsdna”是指双链dna,“bp”是指碱基对,“dntp”是指三磷酸脱氧核苷酸,“rna”是指核糖核酸,“trna”是指转运rna,“rrna”是指核糖体rna,“sirna”是指小干扰rna,“mirna”是指微rna,“mrna”是指信使rna以及“cdna”是指互补dna。在本申请的上下文中的术语“accucal-p”是指用任何合适的荧光团标记的通用参照核酸。在本发明的上下文中的术语“accucal-d”是指使用任何合适的荧光嵌入染料标记的通用参照核酸。在本发明的上下文中的术语“accubeacon”是指用任何合适的荧光团标记的通用参照核酸,其中核酸形成发夹结构。本发明的优选实施方式本发明的动机是由于缺乏精确和有效的手段用于定量对照和处理动物/人类组中的核酸表达。其还激励于许多已知的基因表达研究中所使用的参照基因应答于实验条件或处理而改变表达水平,从而使结果偏移的事实。本发明的优点在于所描述的方法去掉了对于用于标准化数据和定量基因表达的参照基因或合成参照基因的需要。在本文描述的新颖方法中,使用已知量的荧光团标记的通用参照核酸产生校准曲线。通过系列稀释荧光团标记的通用参照核酸并绘制相对于荧光团标记的参照核酸浓度的荧光水平来产生校准曲线。不需要扩增通用参照核酸来产生校准曲线。这与用于评估基因表达的现有方法形成对比,其中以并列的方式或在一个反应混合物中以组合的方式对测试样品和参照基因/合成参照基因两者进行扩增。一旦制备出相同的校准曲线,如果需要定量超过一种的靶核酸,可以多次使用校准曲线。用于制备校准曲线的荧光团标记的通用参照核酸随时间变化是稳定的,例如在约一个月的时间内,以及将溶液反复冷冻和解冻。这使得能够在任何实验要求之前准备和储存荧光团标记的通用参照核酸溶液。在本发明的方法中使用的荧光团标记的通用参照核酸不是所描述的“参照基因”或“合成参照基因”,即其不需要与靶核酸一起扩增。荧光团标记的通用参照核酸不需要与靶核酸或任何参照基因序列具有任何同源性。然而,由于在本发明的方法中使用荧光团标记的通用参照核酸的特定方式(即,在将荧光团标记的通用参照核酸系列稀释之后创建校准曲线),该荧光团标记的通用参照核酸可以具有一定程度的同源性或甚至可以与靶核酸序列或参照基因或其较小部分相同。用限定数量的荧光团(通常为1个)标记每个通用参照核酸。类似地,用已知数量的荧光团标记探针,该荧光团允许由荧光团标记的通用参照核酸所产生的荧光与由荧光团标记的探针结合的靶核酸所产生的荧光进行直接比较。因此,荧光团标记的通用参照核酸的长度和/或组成是无关紧要的,只要可以测定到限定量的荧光团即可。在本文所述的实施方式中,荧光团标记的参照核酸的长度为21bp,并且其gc含量在30%-80%之间变化,但是本领域技术人员应理解,任何长度或gc含量的荧光团标记的参照核酸将提供相似的结果。如果从生物学来源、天然的或其它来源获得荧光团标记的通用参照核酸,则可以使用限制酶以获得合适的片段。也可以方便地使用合成的荧光团标记的通用参照核酸,并且可以通过已知技术简单地制备,如例如sambrook等人,molecularcloning:alaboratorymanual,第二版(1989)coldspringharborlaboratory出版,coldspringharbor,n.y.以及roe等人,dnaisolationandsequencing”(基础技术系列)(1996)johnwiley&sons,inc.,n.y中所描述的那些。可以将相同的荧光团标记的通用参照核酸用于许多核酸靶标。可替换地,可以使用不同的荧光团标记的通用参照核酸和同样不同的荧光团标记的探针来产生多个校准曲线以在多重pcr反应中定量核酸。因此,由于本发明的方法满足了扩增参照基因并且不断地进行参照基因或合成参照基因测定的需要以在每个实验中将核酸表达数据标准化,该方法是有利的。可以制备一种校准曲线并将其用于超过一种的可以在大小和序列上变化的靶核酸的定量,从而与常规基因表达测定相比缩减了成本和时间。作为用于制备本发明方法中使用的校准曲线的通用指南,采用与用于靶核酸和相同反应管一样的反应缓冲液一式两份地将荧光团标记的通用参照核酸在一定浓度范围内系列稀释,并使其经受与靶核酸相同的扩增条件。现在将参考特定但非限制性的描述特定组成和使用方法的实施例来更加详细地描述本发明。然而,应当理解,所包括的特定步骤、组成和方法的详细描述仅仅是为了举例说明本发明。不应该以任何方式理解为是对上述发明观点的广泛描述的限制。实施例实施例1:使用accucal-p定量一系列经过扩增的λdna。将fam标记的参照核酸(21bp和30%gc-accucal-p)稀释至0nm、20nm、40nm、60nm、80nm、100nm和120nm,并且测量40次pcr循环内的荧光(图1a)。通过绘制每次循环的荧光值相对于参照核酸的浓度来创建校准曲线,并计算线性回归(图1b)。在40次pcr循环内扩增一系列的10倍系列稀释的原种λdna,其中使用fam标记的水解探针检测92bp片段的经过扩增的λdna(图1c)。以一式三份进行每个pcr。与阴性对照(ntc)一起,接种核酸的理论量在4.5×107至4.5×101拷贝数/pcr的范围内。使用校正曲线以及每个反应在扩增曲线的指数部分上的各自效率,使用公布的算法来定量接种到每个扩增反应中的核酸的初始量(tichopad等人.2003,nucl.acidsres.,31(20):e122)。由校准曲线,测定每个扩增反应在扩增曲线的指数部分内的dna的pmol,使用方程式pmz=pm/en计算初始输入dna平均值,其中pmz是在时间为零时的pmol,e为效率,n是循环数。使用pmz×6.02223×10-12将pmz转换成拷贝数/pcr。对于每个一份三份,测定初始核酸的平均量以及平均值的标准误差(表1)。接种的理论量(c/pcr)计算的平均量(c/pcr)平均值的标准误差4.5×1014.59×1011.22×1014.5×1021.91×1023.95×1014.5×1033.87×1031.11×1034.5×1041.86×1044.51×1034.5×1056.17×1051.56×1054.5×1061.01×1072.67×1064.5×1077.89×1071.88×107ntc00表1实施例2:使用accucal-p定量一系列经过扩增的λdna。将fam标记的参照核酸(21bp和40%gc-accucal-p)稀释至0nm、20nm、40nm、60nm、80nm、100nm和120nm,并且测量40次pcr循环内的荧光(图2a)。通过绘制每次循环的荧光值相对于参照核酸的浓度来创建校准曲线,并计算线性回归(图2b)。在40次pcr循环内扩增一系列的10倍系列稀释的原种λdna,其中使用fam标记的水解探针检测92bp片段的经过扩增的λdna(图2c)。以一式三份进行每个pcr。与阴性对照(ntc)一起,接种核酸的理论量在4.5×107至4.5×101拷贝数/pcr的范围内。使用校正曲线以及每个反应在扩增曲线的指数部分上的各自效率,使用公布的算法来定量接种到每个扩增反应中的核酸的初始量(tichopad等人.2003,nucl.acidsres.,31(20):e122)。由校准曲线,测定每个扩增反应在扩增曲线的指数部分内的dna的pmol,使用方程式pmz=pm/en计算初始输入dna平均值,其中pmz是在时间为零时的pmol,e为效率,n是循环数。使用pmz×6.02223×10-12将pmz转换成拷贝数/pcr。对于每个一份三份,测定初始核酸的平均量以及平均值的标准误差(表2)。接种的理论量(c/pcr)计算的平均量(c/pcr)平均值的标准误差4.5×1016.51×1011.23×1014.5×1022.95×1027.79×1014.5×1039.07×1032.50×1034.5×1041.89×1044.50×1034.5×1058.66×1052.76×1054.5×1061.43×1073.71×1064.5×1079.68×1072.34×107ntc00表2实施例3:使用accucal-p定量一系列经过扩增的λdna。将fam标记的参照核酸(21bp和62%gc-accucal-p)稀释至0nm、20nm、40nm、60nm、80nm、100nm、120nm、140nm、160nm和200nm,并且测量40次pcr循环内的荧光(图3a)。通过绘制每次循环的荧光值相对于参照核酸的浓度来创建校准曲线,并计算线性回归(图3b)。在40次pcr循环内扩增一系列的10倍系列稀释的原种λdna,其中使用fam标记的水解探针检测92bp片段的经过扩增的λdna(图3c)。以一式三份进行每个pcr。与阴性对照(ntc)一起,接种核酸的理论量在4.5×107至4.5×101拷贝数/pcr的范围内。使用校正曲线以及每个反应在扩增曲线的指数部分上的各自效率,使用公布的算法来定量接种到每个扩增反应中的核酸的初始量(tichopad等人.2003,nucl.acidsres.,31(20):e122)。由校准曲线,测定每个扩增反应在扩增曲线的指数部分内的dna的pmol,使用方程式pmz=pm/en计算初始输入dna平均值,其中pmz是在时间为零时的pmol,e为效率,n是循环数。使用pmz×6.02223×10-12将pmz转换成拷贝数/pcr。对于每个一份三份,测定初始核酸的平均量以及平均值的标准误差(表3)。接种的理论量(c/pcr)计算的平均量(c/pcr)平均值的标准误差4.5×1018.58×1013.06×1014.5×1026.22×1021.15×1024.5×1037.48×1031.48×1034.5×1046.42×1041.26×1044.5×1054.44×1058.09×1044.5×1061.30×1072.42×1064.5×1079.19×1071.68×107ntc00表3实施例4:使用accucal-p定量一系列经过扩增的λdna。将fam标记的参照核酸(21bp和80%gc-accucal-p)稀释至0nm、20nm、40nm、60nm、80nm、100nm和120nm,并且测量40次pcr循环内的荧光(图4a)。通过绘制每次循环的荧光值相对于参照核酸的浓度来创建校准曲线,并计算线性回归(图4b)。在40次pcr循环内扩增一系列的10倍系列稀释的原种λdna,其中使用fam标记的水解探针检测92bp片段的经过扩增的λdna(图4c)。以一式三份进行每个pcr。与阴性对照(ntc)一起,接种核酸的理论量在4.5×107至4.5×101拷贝数/pcr的范围内。使用校正曲线以及每个反应在扩增曲线的指数部分上的各自效率,使用公布的算法来定量接种到每个扩增反应中的核酸的初始量(tichopad等人.2003,nucl.acidsres.,31(20):e122)。由校准曲线,测定每个扩增反应在扩增曲线的指数部分内的dna的pmol,并且使用方程式pmz=pm/en计算初始输入dna平均值,其中pmz是在时间为零时的pmol,e为效率且n是循环数。使用pmz×6.02223×10-12将pmz转换成拷贝数/pcr。对于每个一份三份,测定初始核酸的平均量以及平均值的标准误差(表4)。接种的理论量(c/pcr)计算的平均量(c/pcr)平均值的标准误差4.5×1011.26×1023.63×1014.5×1023.71×1028.98×1014.5×1037.67×1031.90×1034.5×1043.38×1047.96×1034.5×1055.76×1051.53×1054.5×1061.33×1073.27×1064.5×1078.30×1071.93×107ntc00表4实施例5:使用accucal-d、accucal-p和accubeacon定量一系列经过扩增的λdna。使用三种参照核酸独立地对一系列λdna进行定量:accucal-d(用evagreen标记的21bp和62%gc的参照核酸),accucal-p(用fam标记的21bp和62%gc的参照核酸)和accubeacon(用fam标记的21bp和62%gc的发夹式参照核酸)。将每种参照核酸(accucal-d、accucal-p和accubeacon)稀释至0nm、20nm、40nm、60nm、80nm、100nm、140nm和200nm。由在4.5×106至4.5×102范围内的10倍系列稀释的λdna以及ntc扩增92bp片段,并且使用fam标记的水解探针(图5a)或嵌入染料evagreen(图5b)进行检测。使用accucal-d、accucal-p或accubeacon作为校准物计算初始核酸输入量,并且将其与接种至每个pcr中的核酸的理论量进行比较(表5a和5a)。表5a表5a实施例6:使用accucal-d或accucal-p定量一系列扩增子大小。设计引物以扩增来自λdna的92bp、501bp和1000bp的扩增子(分别地,图6a、6b和6c)。由在4.5×105至4.5×101范围内的10倍系列稀释的λdna以及ntc扩增每个片段。使用accucal-p(用fam标记的21bp和62%gc的参照核酸)或accucal-d(用evagreen标记的21bp和62%gc的参照核酸)作为校准物计算初始核酸输入量(表6)。表6实施例7:使用accucal-p定量不同组成的核酸。设计合成靶核酸,由此可以扩增为75%cg组成(图7a)或75%at组成(图7b)的90bp扩增子。将合成模板稀释10倍以使得每个pcr接种1×107至1×103范围内的已知量的模板以及ntc。将accucal-p(用fam标记的21bp和62%gc的参照核酸)稀释至0nm、20nm、40nm、80nm、100nm和120nm。使用accucal-p作为富含gc的模板(表7a)和富含at的模板(表7b)的校准物来计算初始核酸输入量。表7a表7b实施例8:可以同时在同一孔中检测不同量的分别用不同荧光团标记的四种参照核酸的荧光分别地,用fam(图8a)、hex(图8b)、德克萨斯红(图8c)和tye665(图8d)标记参照核酸(21bp和62%gc),并以绿色、黄色、橙色和红色滤波器通道进行检测。在每种情况下,将荧光团标记的参照核酸稀释至0nm、20nm、40nm、60nm、80nm、100nm和160nm,并且在每个pcr扩增循环的退火步骤结束时测量荧光。针对四种参照核酸中的每一种,产生校准曲线(图8e至图8h)。绘制各个浓度的校准物的每次循环的荧光值,并计算线性回归。在每种情况下,显示线性回归线的方程式和r2值。0nm校准物代表试剂、塑料等的荧光的背景水平,并从所有值中将其去除,以提供各个浓度的校准物的真正荧光值。使用稀释为0nm、20nm、40nm、60nm、80nm、100nm、120nm、140nm、160nm和200nm并用fam、hex或德克萨斯红标记的三种参照核酸(21bp和62%gc)来定量一系列的λpcr(表8)。使用fam标记的参照核酸来定量用fam标记的水解探针检测的λpcr。使用hex标记的参照核酸来定量用hex标记的水解探针或joe标记的水解探针检测的λpcr。使用德克萨斯红标记的参照核酸来定量用德克萨斯红标记的水解探针或alexa594标记的水解探针检测的λpcr。表8实施例9:使用以一种荧光团标记的参照核酸来定量使用不同荧光团检测的扩增。扩增λdna(4.5×105和4.5×104c/pcr),并且使用fam标记的水解探针(图9a)、hex-(黑)或joe-(灰)标记的水解探针(图9b)、alexa594-(黑)或德克萨斯红(灰)标记的水解探针(图9c)或cy5-(黑)或tye665-(灰)标记的水解探针(图9d)进行检测。将fam标记的参照核酸(21bp和62%gc)稀释至0nm、20nm、40nm、60nm、80nm、100nm、120nm和140nm,并用于定量所有的扩增(表9)。表9实施例10:accucal-p和样品两者都可以是复合的。将用fam、hex或德克萨斯红(accucal-p)标记的三种参照核酸(21bp和62%gc)各自稀释至0nm、20nm、40nm、60nm、80nm、100nm、120nm、140nm、160nm和200nm,并将其合并在pcr板的一个孔中。在其它孔中,扩增4.5×106至4.5×103c/pcr范围内的十倍稀释的λdna以及ntc。扩增每个长度为约100bp的不同的λ扩增子,并且单独使用fam、hex或德克萨斯红标记的探针检测(曲线显示为黑色),或者所有探针在一起在同一孔中进行检测(曲线显示为灰色),并以绿色(fam)通道(图10a)、黄色(hex)通道(图10b)或橙色(texasred)通道(图10c)检测。使用具有相同荧光团的accucal-p参照核酸计算dna输入量(表10)。表10实施例11:accucal-p可用于定量mirna。使用以fam标记的水解探针的mirna试剂盒(appliedbiosystems)从大鼠脑rna扩增hsa-mir-34a。逆转录后,对rna进行1:3.5的稀释,与无反转录的对照(输入rna,图11中的浅灰色)和ntc(无cdna,但含有所有的其它试剂;图11中的浅灰色)一起,同时将纯的(图11中的深灰色)和经过稀释的(图11中的黑色)cdna以一式三份扩增。将fam标记的参照核酸(accucal-p)稀释至0nm、20nm、40nm、60nm、80nm、100nm、120nm、140nm、160nm和200nm,并用于测定核酸输入量(表11)。样品计算的平均量(c/pcr)样品14.42×105样品21.32×105无反转录0ntc0表11虽然已经通过实施例描述了本发明,但是应当理解,在不脱离本发明的范围下,可以进行变化和修改。此外,如果存在针对于特定特征的已知等同物,则如同特定地引用而将其并入本说明书中。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1