融合笼:用于选择性俘获多环芳烃的紫罗碱类吡啶嗡基笼的合成的制作方法
本申请根据35U.S.C.119要求申请序号为62/045,511、2014年9月3日递交的、名称为“融合笼:用于选择性俘获多环芳烃的紫罗碱类吡啶嗡基笼的合成”的美国临时专利申请的优先权,其全部内容在此并入本申请作为参考。
技术领域
本发明公开涉及用于俘获多环芳烃的吡啶嗡基笼。
背景技术:
相关技术描述
多环芳烃(PAH化合物,分子由两个或多个融合的芳环构成的)通常在天然原油沉积物中被发现,也在碳基材料的不完全燃烧过程过程的人为方法中产生。PAH的致癌特性已被知晓很久了,它们导致诱变发生的路径被很好地归档了。它们不仅在环境中较为普遍,而且是因为它们在水中的低溶解度使得它们也很持久。但是较小的PAH如萘具有稍高一点的水溶解度,因此容易浸出进入到水道之内。尽管有这些情况,并且暗示其与几种疾病状态相关,但是萘每年都以较大的规模被制造。尽管已经报导了许多宿主基于分散力和疏溶剂的效果对PAH有亲合力,受体的相互作用已经开始干扰π电子不足的宿主,从而导致甚至在有机溶剂中对PAH的更高的约束亲合力。迄今为止,几乎不存在分子化合物能从环境中除去PAH。
N,N'-二氮杂大双环聚醚(穴醚)是冠醚的三维类似物。该N,N'-二氮杂大双环聚醚结合1A族和IIA族的金属阳离子如此之强以致它们的1:1 配合物作为穴状化合物变得已知1。虽然从冠醚到穴醚的进程迅速地发生时,曾经花费了许多年时间将球瑗2、分子监狱3和半分子监狱4,5更加高度地设计成使其入口成为具有凹内表面的宿主,所述凹内表面供给离子和具有分散的结合点的中性分子形式的客体的配合以收敛的识别点。这些早期的对于宿主-客体化学的开发为构型包括全部有机到金属配位范围的笼形宿主分子的设计与合成打下了基础。这些落入分子笼的伞下的非自然分子出于许多不同的理由已经被设计和合成出来,包括(i)探索和开发它们的几何学,(ii)研究它们作为分子磁体的性质,(iii)在生物医药领域将它们用作分子媒介,和(iv)使用它们调节和促进化学反应。
申请人报道了环双(百草枯-对-亚苯基)(CBPQT4+;图1:式(I))的更高同系物的有效模板引导合成,通过在两个吡啶鎓环之间阶梯式地插入对亚苯基环来延伸双吡啶鎓单元,从而制备式(II)的延伸的四阳离子二苯撑环烷烃,这里表示为ExnBox4+(其中n=0-3,图1:式(II))。该式(II)的四阳离子二苯撑环烷烃的n=1时这里表示为ExBox4+(图1:式(IIA))。这些结构作为“二维的箱子”对于PAH的结合很弱,因此需要寻求新的对PAH具有更大的结合亲合力的化合物。
技术实现要素:
在第一方面,提供一种选自式(I)和式(II)的化合物:
所述化合物包括与合适的对阴离子形成的盐。
在第二方面,提供一种从包含多环芳烃(PAH)化合物的溶剂分离PAH化合物的方法。该方法包括将包含PAH化合物的溶剂与式(I)或式(II)化合物或其结合接触的步骤:
所述化合物包括与合适的对阴离子形成的盐。
在第三方面,提供一种从溶剂混合物分离多环芳烃(PAH)化合物的方法。该方法包括几个步骤。第一步包括使溶剂混合物与分离媒质接触。所述分离媒质由选自式(I)、式(II)的化合物或其结合物构成:
所述化合物包含与合适的对阴离子形成的盐。第二步包括使用液相色谱移动相从溶剂混合物解析PAH化合物。第三步包括从溶剂混合物分离PAH化合物。
本发明的这些以及其他特征、目标和优点可以从如下的说明中得到更好的理解。在本说明书中,附图可以作为参考,其构成发明的一部分,以解释说明、非限制性的实例的方式显示。
附图说明
在考虑下面的详细说明时这些除了上面提到的特征、目标和优点将变得更容易清晰可见。这些详细说明提到了以下的附图。
图1描述了三个示例性的现有技术化合物,具有式(I)、(II)和(IIA)。
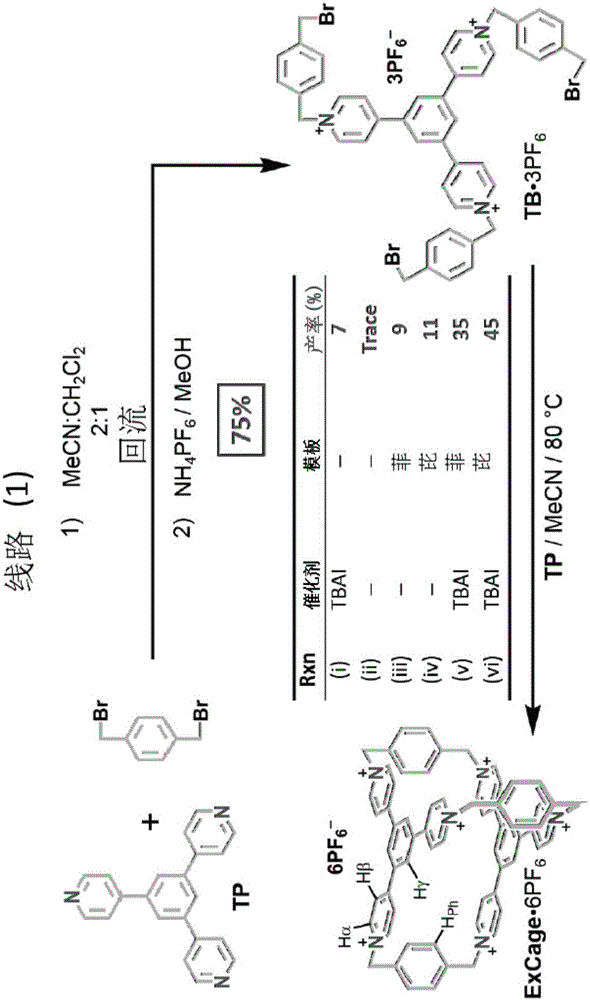
图2描述了合成融合笼(ExCage)6PF6的示例性流程图。
图3描述了合成蓝笼(BlueCage)6PF6的示例性流程图。
图4的(a)示例性地描述了当萘的水饱和溶液包含少量的己-2,5-二酮作为内标时GC/MS扫描显示的结果。
图4的(b)示例性地描述了在加入ExCage6+作为它的不溶PF6-盐、超声处理、滤出固体并将溶液注入到GC/MS内从而揭示仅含内标物质,例如所有的萘已经从饱和水溶液中清除的GC/MS扫描显示结果。
图4的(c)示例性地描述了当那些被滤出的固体(上图)被溶解于MeCN并注入到GC/MS中时,例如萘的峰再现和没有内标物质的GC/MS扫描显示结果。
图5描述了在MeCN中ExCage6+和存在于八个PAH(从平面性引入了前述的偏离)、苯并蒽、的π-电子数之间的约束亲合力(log Ka’s)的线性图,八个PAH、苯并蒽、具有延长的结构,位于线以下。
图6示例性地描述了苯并蒽的形成的ITC数据。
虽然本发明可以进行各种改性和替代方式,示范性的实例在附图中举例说明和在此详细描写。但是应该理解那些示范性实例的说明并不意味着将本发明限制为公开的特定形式,相反地本发明旨在覆盖所有落入如所述具体实例和权利要求限定的本发明精神和范围的改进、替代和备选方案。
因此实例参考和权利要求被用于解释本发明的范围。
具体实施方式
下文中将要更充分地参考附图描述所述组合物和方法,其中将本发明的实例的部分而非全部排列和变化示出了。实际上,本发明可以体现为许多不同的形式,而不应被认为限制为这里阐述的实例。这些实例以充分的撰写细节进行描述并使得本领域技术人员能够制造和使用本发明,一起公开的还有用于实现本发明的最佳方式,如权利要求及其等价物所定义的那样。
同样地,这里描述的组合物和方法的诸多改变和其他实例会被本领域技术人员基于上述说明书和附图中存在的教导而想到。因此,将理解本发明不限于公开的特定实例,并且意图将那些改进和其他实例包括在权利要求的范围内。尽管这里使用了专用名词,它们只是用作普通和叙述的功能,不为了作限制。
除非另外定义,这里使用的所有技术和科学名词就本发明所属的含义具有和本领域技术人员通常理解相同的意思。
尽管任何与这里描述的那些相似或等价的方法和材料可以被用于本发明的实践或测试,优选的方法和材料如下所述。
此外,用不定冠词“a”或“an”表示的要素并不排除存在多于一个(单数)的可能性,除非上下文明确地要求只有一个要素。不定冠词“a”或“an”由此通常指“至少一个”。
这里使用的“约”意味着一个数值的统计学意义的范围,例如规定的浓度、长度、分子量、pH值、序列同一性、时间范围、温度或体积。这样的数值或范围可以在一个数量级内,典型的在给定值或范围的20%内,更典型的在10%内,进而更典型的在5%内。通过“约”包含的允许偏差取决于研究中的特定系统,能容易地被本领域技术人员所理解。
概述
本申请人产生了一个基本的发现,即对于PAH具有改善的约束亲合力的两种新化合物。这些化合物的优点包括:(a)对于PAH比类似的二维的箱结构具有两倍或者更高量级大小的约束力;(b)阴离子交换允许所 述化合物在固态下、或溶解在水中或有机媒质中起作用;和(c)对PAH的约束有选择性。这些化合物的应用包括:(a)过滤/隔离PAH到从人为起源低至两个融合芳环的程度;(b)从有机媒质俘获PAH;(c)从空气中过滤PAH;(d)从水媒质俘获PAH;(e)从生物学上重要的分子的复杂混合物过滤/隔离PAH;和(f)以比色的方式检测大量的PAH。
组合物、合成方法和用作PAH化合物结合媒质
在第一方面,通过将ExBox4+的中央1,4-二取代的苯环型环结构改变为1,3,5-三取代的结构来提供式(I)的二环六阳离子二苯撑环烷烃(这里命名为ExCage6+):
在第二方面,通过将ExBox6+的中央1,3,5-三取代的苯环型环结构改变为1,3,5-三取代的三嗪环来提供式(II)的二环六阳离子二苯撑环烷烃(这里命名为BlueCage6+):
ExCage·6PF6的合成(图2的路线1)始于1,3,5-三(4-吡啶基)苯(TP),TP在MeCN/CH2Cl2(2:1)中用15倍过量的1,4-双(溴甲基)苯进行烷基化,回流3天,以75%的产率提供TB·3PF6的三溴化物,接着在MeOH中进行平衡离子交换(NH4PF6)。该三溴化物与TP在MeCN中的另一等价物在作为催化剂30的0.3当量的碘化四丁铵(TBAI)存在下在80℃反应36小时,提供粗的ExCage·6Cl,接着添加TBACl到反应混合物中以沉淀粗产品,该粗产品在准备的反相高效液相色谱法之后从洗脱液用NH4PF6沉淀,以7%的产率获得ExCage·6PF6。在没有催化剂的情况下仅有痕量的ExCage·6PF6被分离。但是,当在催化剂存在下重复该反应时,首先使用菲(6当量)作为模板,然后用芘(6当量),产率得到很大改进。在两个TBAI存在的模板直接合成中,随后添加TBACl的反应混合物必须经受连续地使用CHCl3的液-液萃取以除去模板,该萃取在经受预备的反相色谱法之前,随后通过添加NH4PF6到洗脱液中进行平衡离子交换。尽管使用芘作为模板能将ExCage·6PF6的产率提高到45%,但是模板被证明有点难以通过连续的液-液萃取来除去,而菲更容易用CHCl3萃取,其最终产品的产率为35%。在没有催化剂存在下但是模板存在下,在室温下进行21天后反应的产率是相当低的,即对于使用菲和芘分别为9和11%。
通过一个与上述用于ExCage·6PF6相似的步骤(图3的路线2)来实现模板直接合成BlueCage·6PF6。用10当量的1,4-双(溴甲基)苯在MeCN/CH2Cl2(1:1)处理2,4,6-三(4-吡啶基)-1,3,5-三嗪(TPT),在90℃下加热3天,以58%的产率提供TPTB,接着平衡离子交换。在6当量作为模板的菲和20摩尔%的碘化四丁铵(TBAI)在MeCN中的存在下,TPTB·3PF6与TPT在90℃下反应3天,供给菲·6Br,接着用溴化四丁铵(TBABr)沉淀粗产品。用CHCl3连续萃取7天,接着进行预备的反相高效液相层析法和用含水的NH4PF6进行阴离子交换,以11%的产率提供作为白色固体的BlueCage·6PF6。
在一个方面,提供一种从包含PAH化合物的溶剂分离PAH化合物的分离媒质。该分离媒质包含选自式(I)和(II)或其结合的二环六阳离子二苯撑环烷烃。
在另一个方面,提供从包含PAH化合物的溶剂分离PAH化合物的方法。参考图4,所述方法包括将溶剂与式(I)或(II)或其结合接触的步骤,所述溶剂包含PAH化合物。优选,式(I)或(II)化合物或其结合被配置作为静止相,其中所述静止相与所述包含PAH化合物的溶剂接触。所述溶剂可以是水性的或有机溶剂。所述包含式(I)或(II)的化合物的静止相优选被配置在用于进行从包含PAH化合物的溶剂色谱分离PAH化合物的柱中。优选的用于该目的的色谱分离包括高效液相色谱法(HPLC)。因为PAH化合物通常是疏水性,优选的层析媒质包括疏水性溶剂如己烷。
优选的用于分离的PAH化合物包括任何二维的多环芳烃。示例性的能够遵照与式(I)和(II)化合物具有高度亲合力结合的PAH化合物包括萘、菲、苯并蒽、芘、螺烯、三亚苯、二萘嵌苯和六苯并苯(图5)。式(I)和(II)的化合物与PAH化合物形成1:1化学计量的复合物(例如参见图6)。在式(II)化合物的情况下,阴离子的类型可以调节所述化合物对PAH化合物的约束。例如,PF6-阴离子减轻了PAH化合物结合到式(II)化合物的程度,由此导致在包含PF6-的溶液中相比式(I),PAH化合物对式(II)化合物的结合亲合力更低。该影响可以通过交换大的阴离子如BArF-阴离子来代替包含式(II)化合物的溶液中的PF6-阴离子而被反转,其中式(II)化合物显示对给定的PAH化合物与式(I)可比拟的约束亲合力。
式(I)和(II)化合物可以被用于以比色的方式检测大量的PAH。通过比色的性质来检测PAH的证据可以从单晶化合物-PAH复合物的颜色看出,例如那些被用于通过X-射线衍射检测复合物构造。例如,当ExCage是无色的,用苯并蒽、芘和二萘嵌苯配合获得的晶体是黄色、橙色和红色的。这种颜色变化也可以在结合ExCage和特定的PAH的瞬间在溶液中被观察到。
本发明要求保护的主体不会以任何方式被束缚或限制任何的特别理论、高结合亲合力的机理,用式(I)或(II)的化合物保留PAH化合物是部分地基于其三维的二环结构以及所述化合物的π-电子不足。
应用
在第一方面,提供一种选自式(I)和式(II)的化合物:
所述化合物包含与合适的对阴离子形成的盐。
在第一方面的第一点,所述化合物由式(I)的物质构成:
在第一方面的第二点,所述化合物由式(II)的物质构成:
在第一方面的第三点,所述合适的对阴离子选自PF6-和BArF-。
在第二方面,提供一种根据上述第一方面的化合物构成的分离媒质。其中,所述分离媒质包括选自PF6-和BArF-的合适的对阴离子。
在第三方面,提供一种从包含多环芳烃(PAH)化合物的溶剂分离PAH化合物的方法。该方法包括将包含PAH化合物的溶剂与式(I)或式(II)化合物或其结合接触的步骤:
所述化合物包括与合适的对阴离子形成的盐。
在第一方面,所述方法包括具有式(I)结构的化合物:
或者,所述方法包括具有式(II)结构的化合物:
这其中,所述化合物包括选自PF6-和BArF-的合适的对阴离子。这其中,所述溶剂选自水的或有机溶剂。这其中,所述PAH化合物由平面PAH化合物构成。所述PAH化合物选自由萘、菲、苯并蒽、芘、螺烯、三亚苯、二萘嵌苯和六苯并苯或其结合构成的组。
在第四方面,提供一种从溶剂混合物分离多环芳烃(PAH)化合物的方法。该方法包括几个步骤。第一步包括使溶剂混合物与分离媒质接触。第二步包括使用液相色谱移动相从溶剂混合物解析PAH化合物。第三步包括从溶剂混合物分离PAH化合物。所述分离媒质由选自式(I)、式(II)的化合物或其结合物构成:
所述化合物包含与合适的对阴离子形成的盐。其中,所述方法包括一种化合物,其中合适的对阴离子选自PF6-和BArF-。进而,所述PAH化合物选自由萘、菲、苯并蒽、芘、螺烯、三亚苯、二萘嵌苯和六苯并苯或其结合构成的组。进而,所述液相色谱移动相包括己烷。进而,所述分离媒质放置在色谱柱中。其中,所述色谱柱设置为高效液相色谱。
实施例
在考虑下列非限制性的实施例的基础上将更充分地理解本发明,该实施例用于解释的目的,并不构成发明的限制。
实施例1:材料和方法
所有的试剂从商业供应商处购得,除非另有说明不需要进行进一步的纯化而使用。如之前文献中报道地那样合成2,4,6-三(4-吡啶基)-1,3,5-三嗪8和2,4,6-三(苄基)吡啶鎓-4-基)-1,3,5-三嗪三(六氟磷酸盐)9。溶剂通过Ar来脱氧30分钟。如之前报道10的那样合成1,3,5-三(4-吡啶基)苯。使用C18柱和二元溶剂系统(MeCN和带有0.1%CF3CO2H的水)、在反相HPLC(RP-HPLC)装置上进行分析的高效液相色谱(HPLC)。使用UV-3600岛津分光光度计记录紫外-可见光-近红外吸收光谱。在BrukerAvance 600、Varian P-Inova 500、和Bruker F500分光仪上记录核磁共振(NMR)谱,工作频率分别为600、500、和500MHz(1H NMR),以及150、125、和125MHz(13C NMR)。化学位移是以相对于剩余的非氘化溶剂(CD3CN:δH=1.94ppm,δC=1.32,和118.26ppm;DMF-d7:δH=8.03, 2.92,和2.75ppm,δC=29.76,34.89,和163.15ppm)的信号的ppm(百万分之一)进行记录。在Agilent 6210飞行时间(TOF)LC-MS上、使用用Agilent 1100 HPLC堆栈偶联的ESI源,测定高分辨质谱(HRMS),直接注入(0.6mL/min)。在MicroCal系统、VP-ITC模型上进行等温滴定量热法(ITC)试验。在1.8mL的室内使用ExCage·6PF6在MeCN中的溶液作为宿主溶液。通过连续注射10μL的滴定标准液加入PAH在MeCN中的溶液,每次注射20秒(25次),每次注射之间间隔300秒。使用单址绑定模型利用数据计算热力学信息,所述数据减去客体的稀释热,报道运行三次的平均值。在室温下、氩气净化DMF溶液中,使用接口连接电脑的Gamry通用工具(Reference 600)进行循环伏安法(CV)实验。使用玻璃碳工作电极(0.071cm2)进行所有的CV实验。电极表面在使用前立即用0.05μm氧化铝水浆料在毡表面上进行常规的抛光。反电极是Pt线圈,参考电极是Ag/AgCl电极。样品和支持电解质(四丁基铵六氟磷酸盐(TBAPF6))的浓度分别为1.0mM和0.10M。CV单元在使用前立即在烤箱中干燥,并且在该单元的温度冷却到室温时不断地用氩气充满该单元,以避免水冷凝。
实施例2.用于制备式(I)化合物的合成方法
图2中所示的路线(1)提供了一种式(I)化合物的合成的概览。
1)1,3,5,-(1-甲基吡啶4-基)苯三(六氟磷酸盐)—TM·3PF6
TM·3PF6的合成是根据路线(1A)来实现的:
(路线(1A))
TM·3PF6:将1,3,5-三(4-吡啶基)苯(25.0mg,0.08mmol)和MeI(150μL,2.40mmol)在MeCN(5.0mL)中的混合物在50℃、搅拌下加热18小时。将反应混合物冷却到室温,通过过滤收集沉淀。所得固体溶解在MeOH(50mL)中,接着添加NH4PF6,在50mL水中稀释,得到的纯TM·3PF6(55.5mg,87%)沉淀通过过滤进行收集,为无色固体。TM·3PF6的HRMS-ESI;C24H24Br3F12N3P2的计算值:m/z=644.1249[M–PF6]+;实验值:644.1262[M–PF6]+。1H NMR(500MHz,CD3CN,ppm):δH 8.77(AA'XX'的AA',J=6.9Hz,6H),8.53(s,3H),8.45(AA'XX'的XX',J=7.0Hz,6H),4.36(s,9H.13C NMR(125MHz,CD3CN,ppm):δC 154.8,146.6,137.7,131.6,126.6,48.9。
2)1,3,5,-(1-(4-溴代甲基苄基)吡啶鎓-4-基)苯
TB·3PF6的合成是根据路线(1B)来实现的:
(路线1B)
三(六氟磷酸盐)—TB·3PF6
TB·3PF6:将α,α'-二溴-对二甲苯(8.96g,33.9mmol)加入到MeCN/CH2Cl2(1:1v/v,113mL)中,悬浮液被加热到60℃直到所有化合物均溶解。将溶液的温度升高到90℃,1,3,5-三(4-吡啶基)苯(TP)(700mg,2.26mmol)在MeCN(37mL)中的悬浮液等份数加入,历经2小时。在回流下加热3天后,将反应混合物冷却到室温,沉淀在CH2Cl2(500mL)中稀释并通过过滤收集。所得沉淀溶解在MeOH(100mL)中,接着添加过量的NH4PF6在水(400mL)中的混合物,得到纯的TB·3PF6(2.19g,75%)沉淀, 过滤收集沉淀,为无水固体。1H NMR(500MHz,CD3CN,ppm):δH 8.90(AA'XX'的AA',J=6.9Hz,6H),8.53(s,3H),8.49(AA'XX'的XX',J=6.9Hz,6H),7.55(AA'BB'的AA',J=8.3Hz,6H),7.49(AA'BB'的BB',J=8.2Hz,6H),5.78(s,6H),4.61(s,6H)。13C NMR(125MHz,CD3CN,ppm):δC155.9,145.8,141.3,137.7,134.1,132.0,131.1,130.5,127.4,64.6,33.5。TB·3PF6的HRMS-ESI;计算值:C45H39Br3F12N3P2:m/z=1149.9952[M–PF6]+;实验值:1149.9959[M–PF6]+。
3)环双(1,3,5,-三(1,1'-(1,4-亚苯基双(亚甲基))-吡啶鎓-4-基)苯)-六(六氟磷酸盐)—ExCage·6PF6
ExCage·6PF6的合成是根据路线(1C)来实现的:
(路线(1C))
ExCage·6PF6:使用下述不同设置的条件进行六个反应:(i)没有模板,有催化剂,(ii)没有催化剂,没有模板,(iii)没有催化剂,使用菲作为模板,(iv)没有催化剂,使用芘作为模板,(v)有催化剂,使用菲作为模板,和(vi)有催化剂,使用芘作为模板。
(i)没有模板,有催化剂。在80℃下加热TB·3PF6(150mg,0.129mmol)、TP(39.7mg,0.129mmol),和碘化四丁铵(TBAI,14.2mg,0.0383mmol)在干MeCN(50mL)中的溶液36小时。反应通过添加过量的TBACl淬灭,于是从溶液中作为六氯化盐沉淀所述粗产品,将出产品溶解在最小量的H2O/EtOH(19:1,v/v)中,之后经历高效反相C18柱色谱法,以包含0.1%TFA的水作为洗脱液起始,添加至多25%的MeCN/0.1%TFA。该 色谱纯的化合物通过将NH4PF6加入到洗脱液来沉淀,获得纯的ExCage·6PF6(15mg,7%)。1H NMR(500MHz,CD3CN,ppm):δH 8.74(AA'XX'的AA',J=7.0Hz,12H),8.40(s,6H),8.28(AA'XX'的XX',J=6.6Hz,12H),7.57(s,12H),5.73(s,12H)。13C NMR(125MHz,CD3CN,ppm):δC154.2,145.3,136.9,136.7,131.6,131.4,126.5,64.7。用于ExCage·6PF6的HRMS-ESI;计算值C66H54F24N6P4:m/z=755.1483[M–2PF6]2+;实验值:755.1505[M–2PF6]2+。
(ii)没有催化剂,没有模板。将TB·3PF6(150mg,0.129mmol)和TP(39.7mg,0.129mmol)在干的MeCN(50mL)溶液中在室温下搅拌21天。反应通过添加过量的TBACl淬灭,于是从溶液中作为六氯化盐沉淀所述粗产品,将出产品溶解在最小量的H2O/EtOH(19:1,v/v)中,之后经历高效反相C18柱色谱法,以包含0.1%TFA的水作为洗脱液起始,添加至多25%的MeCN/0.1%TFA。该色谱纯的化合物通过将NH4PF6加入到洗脱液来沉淀,获得痕量的纯ExCage·6PF6。
(iii)没有催化剂,使用菲作为模板。将TB·3PF6(150mg,0.129mmol)、TP(39.7mg,0.129mmol)和菲(138mg,0.774mmol)在干MeCN(50mL)中的溶液在室温下搅拌21天。反应通过添加过量的TBACl淬灭,于是从溶液中作为六氯化盐沉淀所述粗产品,溶解在水中,通过使用CHCl3连续液-液萃取3天除去模板。水相被浓缩并溶解在最小量的H2O/EtOH(19:1,v/v)中,之后经历高效反相预备C18柱色谱法,以包含0.1%TFA的水作为洗脱液起始,添加至多25%的MeCN/0.1%TFA。该色谱纯的化合物通过将NH4PF6加入到洗脱液来沉淀,获得纯的ExCage·6PF6(21mg,9%)。
(iv)没有催化剂,使用芘作为模板。将TB·3PF6(150mg,0.129mmol)、TP(39.7mg,0.129mmol)和芘(156mg,0.774mmol)在MeCN(50mL)中的溶液在室温下搅拌21天。反应通过添加过量的TBACl淬灭,于是从溶液中作为六氯化盐沉淀所述粗产品,溶解在水中,通过使用CHCl3连续液-液萃取30天除去模板。水相被浓缩并溶解在最小量的H2O/EtOH(19:1, v/v)中,之后经历高效反相预备C18柱色谱法,以包含0.1%TFA的水作为洗脱液起始,添加至多25%的MeCN/0.1%TFA。该色谱纯的化合物通过将NH4PF6加入到洗脱液来沉淀,获得纯的ExCage·6PF6(26mg,11%)。
(v)有催化剂,使用菲作为模板。将TB·3PF6(150mg,0.129mmol)、TP(39.7mg,0.129mmol)、TBAI(14.3mg,0.039mmol)和菲(138mg,0.774mmol)在干MeCN(50mL)中的溶液在80℃下加热36小时。反应通过添加过量的TBACl淬灭,于是从溶液中作为六氯化盐沉淀所述粗产品,溶解在水中,通过使用CHCl3连续液-液萃取3天除去模板。水相被浓缩并溶解在最小量的H2O/EtOH(19:1,v/v)中,之后经历高效反相预备C18柱色谱法,以包含0.1%TFA的水作为洗脱液起始,添加至多25%的MeCN/0.1%TFA。该色谱纯的化合物通过将NH4PF6加入到洗脱液来沉淀,获得纯的ExCage·6PF6(81mg,35%)。
(vi)有催化剂,使用芘作为模板。将TB·3PF6(150mg,0.129mmol)、TP(39.7mg,0.129mmol)、TBAI(14.3mg,0.039mmol)和芘(156mg,0.774mmol)在干MeCN(50mL)中的溶液在80℃下加热36小时。反应通过添加过量的TBACl淬灭,于是从溶液中作为六氯化盐沉淀所述粗产品,溶解在水中,通过使用CHCl3连续液-液萃取30天除去模板。水相被浓缩并溶解在最小量的H2O/EtOH(19:1,v/v)中,之后经历高效反相预备C18柱色谱法,以包含0.1%TFA的水作为洗脱液起始,添加至多25%的MeCN/0.1%TFA。该色谱纯的化合物通过将NH4PF6加入到洗脱液来沉淀,获得纯的ExCage·6PF6(105mg,45%)。
实施例3.用于制备式(II)化合物的合成方法
图3中示出的路线(2)提供一种合成式(II)化合物的概览。
TPTB·3PF6的合成方法根据路线(2A)来实现:
(路线(2A))
TPTB·3PF6:将1,4-双(溴甲基)苯(7.98g,30.2mmol)在1:1(v/v)MeCN-CH2Cl2(120mL)中的溶液在氩气气氛中加热到90℃,以小比例用固体TPT(703mg,2.25mmol)处理。搅拌所述混合物72小时,经历一段时间生成黄色悬浮液。在冷却到室温后,用CH2Cl2(500mL)稀释悬浮液,通过过滤分离固体。滤饼用大量的CH2Cl2洗涤,然后用MeOH(45mL)打浆。合并的部分被加入到饱和的含水NH4PF6(500mL)中,得到作为白色固体的TPTB·3PF6沉淀(1.70g,1.31mmol,58%)。1H-NMR(500MHz,CD3CN)δ9.25(d,J=6.5Hz,6H),9.08(d,J=7Hz,6H),7.57(d,J=8Hz,6H),7.53(d,J=8.5Hz,6H),5.88(s,6H),4.62(s,6H)。13C-NMR(125MHz,CD3CN)δ170.2,150.5,147.2,141.4,133.5,131.5,130.8,128.6,65.5,33.5.HRMS(ESI)计算值:C42H36Br3F12N6P2:m/z=1152.9809[M–PF6]+;实验值:1152.9816。
BlueCage·6PF6的合成根据路线(2B)来实现:
(路线(2B))
TPTB·3PF6(1.70g,1.31mmol)、2,4,6-三(4-吡啶基)-1,3,5-三嗪(408g,1.31mmol)、TBAI(97mg,0.26mmol)和菲(2.10g,11.8mmol)在MeCN(500mL)中的均匀混合物在氩气气氛中回流下加热3天。反应混合物被冷却到室温,并用过量的TBABr处理以沉淀通过过滤收集的剩余固体。滤饼用最小量的水浆化,合并的水性部分经历使用CHCl3的7天连续萃取,以除去模板。水相在真空中浓缩,用预备的高效液相色谱法纯化,以包含0.1%TFA的水作为洗脱液起始,添加至多25%的MeCN/0.1%TFA。合并部分在真空下浓缩并用饱和的水性NH4PF6处理,提供作为白色固体的纯BlueCage·6PF6(257mg,143μmol,11%)。1H-NMR(500MHz,CD3CN)δ9.06(d,J=6.5Hz,12H),9.05(d,J=6.5Hz,12H),7.63(s,12H),5.82(s,12H)。13C-NMR(125MHz,CD3CN)δ169.4,150.4,146.1,136.8,130.8,128.7,65.7。19F-NMR(470MHz,CD3CN)δ–71.0(d,J=706.9Hz)。31P-NMR(162MHz,CD3CN)δ–143.59(sept,J=708.6Hz).HRMS(ESI)计算值C60H48F24N12P4:m/z=758.1341[M–2PF6]2+;实验值:758.1346。
BlueCage·6PF6用Na[BArF]的阴离子交换以获得BlueCage·6BArF是根据路线(2C)来实现的:
(路线(2C))
BlueCage·6BArF:BlueCage·6PF6(40mg,22μmol)在MeCN(2mL)中的溶液用过量的TBACl处理以沉淀氯化盐,其通过离心分离收集。所述固体用MeCN洗涤很多次,溶解在MeOH中,并用Na[BArF](117mg,132μmol)处理。蒸发所述溶液,剩余物在MeCN中吸收,并通过亚微米过滤器。所述混合物在真空下浓缩,得到的粘性油用最小量的Et2O重复浆化,获得粘性油BlueCage·6BArF(100mg,16.3μmol,74%),静置下凝固。1H-NMR(500MHz,CD3CN)δ9.09(d,J=6.5Hz,12H),8.98(d,J=6.5Hz,12H),7.70(s,12H),7.69(m,48H),7.66(s,24H),5.82(s,12H)。
ExCage·6PF6用Na[BArF]的阴离子交换以获得ExCage·6BArF是根据路线(2D)来实现的:
(路线(2D))
ExCage·6BArF:ExCage·6PF6(40mg,22μmol)在MeCN(2mL)中的溶液用过量的TBACl处理以沉淀氯化盐,其通过离心分离收集。所述固体用MeCN洗涤很多次,溶解在MeOH中,并用Na[BArF](117mg,132μmol)处理。蒸发所述溶液,剩余物在MeCN中吸收,并通过亚微米过滤器。所述混合物在真空下浓缩,得到的粘性油用最小量的Et2O重复浆化,获得粘性油ExCage·6BArF(102mg,16.7μmol,75%),静置下凝固。1H-NMR(500MHz,CD3CN)δ8.78(d,J=7.0Hz,12H),8.38(s,6H),8.28(d,J=6.5Hz,12H),7.69(m,48H),7.59(s,24H),5.75(s,12H)。
将六苯并苯并入到BlueCage·6PF6以获得六苯并苯·6PF6是根据路线(2E)实现的:
(路线2E)
六苯并苯·6PF6:1mg(0.553μmol)BlueCage·6PF6在MeCN(1mL)中的溶液用六苯并苯(2mg,6.66μmol)处理,超声处理1分钟。所述溶液通过亚微米过滤器,并经历iPr2O的蒸气扩散,提供作为深紫色晶体的六苯并苯·6PF6。
参考文献
(1)(a)Dietrich,B.;Lehn,J.-M.;Sauvage,J.-P.Tetrahedron Lett.1969,2889;(b)Dietrich,B.;Lehn,J.-M.;Sauvage,J.-P.Tetrahedron1973,29,1647;(c)Huang,R.H.;Faber,M.K.;Moeggenborg,K.J.;Ward,D.L.;Dye,J.L.Nature 1988,331,599;(d)vonC.;Hampe,O.;Weigend,F.;Stahl,S.Angew.Chem.Int.Ed.2007,46,4775;(e)Rupar,P.A.;Staroverov,V.N.;Baines,K.M.Science 2008,322,1360;(f)Hao,H.-G.;Zheng,X.-D.;Lu,T.-B.Angew.Chem.Int.Ed.2010,49,8148;(g)Lopez,N.;Graham,D.J.;McGuire,R.;Alliger,G.E.;Shao-Horn,Y.;Cummins,C.C.;Nocera,D.G.Science 2012,335,450;(h)Wei,P.;Xia,B.;Zhang,Y.;Yu,Y.;Yan,X.Chem.Commun.2014,50,3973.
(2)(a)Cram,D.J.;Kaneda,T.;Helgeson,R.C.;Brown,S.B.;Knobler,C.B.;Maverick,E.;Trueblood,K.N.J.Am.Chem.Soc.1985,107,3645;(b)Cram,D.J.;Lein,G.M.J.Am.Chem.Soc.1985,107,3657;(c)Bryant,J.A.;Ho,S.P.;Knobler,C.B.;Cram,D.J.J.Am.Chem.Soc.1990,112,5837;(d)Mitjaville,J.;Caminade,A.-M.;Mathieu,R.;Majoral,J.-P.J.Am.Chem.Soc.1994,116,5007;(e)Yi,H.-P.;Wu,J.;Ding,K.-L.;Jiang,X.-K.;Li,Z.-T.J.Org.Chem.2007,72,870;(f)Skowronek,P.;Gawronski,J.Org.Lett.2008,10,4755;(g)Giri,N.;Davidson,C.E.;Melaugh,G.;Del Pópolo,M.G.;Jones,J.T.A.;Hasell,T.;Cooper,A.I.;Horton,P.N.;Hursthouse,M.B.;James,S.L.Chem.Sci.2012,3,2153.
(3)(a)Cram,D.J.;Karbach,S.;Kim,Y.H.;Baczynskyj,L.;Kalleymeyn,G.W.J.Am.Chem.Soc.1985,107,2575;(b)Cram,D.J.;Karbach,S.;Kim,Y.H.;Baczynskyj,L.;Marti,K.;Sampson,R.M.; Kelleymeyn,G.W.J.Am.Chem.Soc.1988,110,2554;(c)Sherman,J.C.;Cram,D.J.J.Am.Chem.Soc.1989,111,4527;(d)Jasat,A.;Sherman,J.C.Chem.Rev.1999,99;(e)Roach,P.;Warmuth,R.Angew.Chem.Int.Ed.2003,42,3039;(f)Makeiff,D.A.;Sherman,J.C.J.Am.Chem.Soc.2005,127,12363;(g)Ihm,C.;Jo,E.;Kim,J.;Paek,K.Angew.Chem.Int.Ed.2006,45,20569;(h)Chen,J.Y.-C.;Jayaraj,Jockusch,S.;Ottaviani,M.F.;Ramamurthy,V.;Turro,N.J.J.Am.Chem.Soc.2008,130,7206;(i)Srinivasan,K.;Gibb,B.C.Chem.Commun.2008,38,4640;(j)Wang,H.;Liu,F.;Helgeson,R.C.;Houk,K.N.Angew.Chem.Int.Ed.2013,52,655.
(4)(a)Cram,D.J.;Tanner,M.E.;Knobler,C.B.J.Am.Chem.Soc.1991,113,7717;(b)Cram,D.J.;Jaeger,R.;Deshayes,K.J.Am.Chem.Soc.1993,115,10111;(c)Makeiff,D.A.;Pope,D.J.;Sherman,J.C.J.Am.Chem.Soc.2000,122,1337;(d)Warmuth,R.;Yoon,J.Acc.Chem.Res.2001,34,95;(e)Warmuth,R.;Makowiec,S.J.Am.Chem.Soc.2007,129,1233;(f)Lu,Z.;Moss,R.A.;Warmuth,R.;Krogh-Jespersen,K.J.Phys.Chem.A 2011,115,13799;(g)Li,M.-J.;Huang,C.-H.;Lai,C.-C.;Chiu,S.-H.Org.Lett.2012,14,6146;(h)Lin,Z.;Sun,J.;Efremovska,B.;Warmuth,R.Chemistry 2012,18,12864.
(5)(a)Cram,D.J.;Tanner,M.E.;Knobler,C.B.J.Am.Chem.Soc.1991,113,7717;(b)Cram,D.J.Nature 1992,356,29;(c)Cram,D.J.;Cram,J.M.容器分子和它们的客体(Container Molecules and their Guests).超分子化学专著;Ed.Stoddart,J.F.英国皇家化学学会,剑桥,1994。当穴醚形成配合物时,它们这些穴状化合物、球瑗、分子监狱和半分子监狱,分别被称为球瑗配合物(spheraplexes)、分子监狱配合物(carceplexes)和半分子监狱配合物
J.C.;M.;Strutt,N.L.;Frasconi,M.;Sampath,S.;Giesener,M.A.;McGrier,P.L;Bruns,C.J.;Stern,C.L.;Sarjeant,A.A.;Stoddart,J.F.J.Am.Chem.Soc.2013,135,183;(b)M.;Barnes,J.C.;Dale,E.J.;Liu,W.-G.;Strutt,N.L.;Bruns,C.J.;Vermeulen,N.A.;Ghooray,K.C.;Sarjeant,A.A.;Stern,C.L.; Botros,Y.Y.;Goddard III,W.A.;Stoddart,J.F.J.Am.Chem.Soc.2013,135,12736;(c)Barnes,J.C.;M.;Vermeulen,N.A.;Dale,E.J.;Stoddart,J.F.J.Org.Chem.2013,78,11962.
(7)(a)Anderson,S.;Anderson,H.L.;Sanders,J.K.M.;Acc.Chem.Res.1993,26,469;(b)Cacciapaglia,R.;Mandolini,L.Chem.Soc.Rev.1993,22,221;(c)Hoss,R.;F.Angew.Chem.Int.Ed.Engl.1994,33,375;(d)Hubin,T.J.;Busch,D.H.Coord.Chem.Rev.2000,200,5;(e)Meyer,C.D.;Joiner,C.S.;Stoddart,J.F.Chem.Soc.Rev.2007,36,1705;(f)Crowley,J.D.;Goldup,S.M.;Lee,A.L.;Leigh,D.A.;McBurney,R.T.Chem.Soc.Rev.2009,38,1530.
(8)Anderson,H.L.;Anderson,S.;Sanders,J.K.M.J.Chem.Soc.Perkin.Trans.1995,2231–2245.
(9)Gries,W.-K.;Günther,E.;Hünig,S.Liebigs Ann.Chem.1991,1021–1028.
(10)Schmittel,M.;He,B.;Mal,P.Org.Lett.2008,10,2513–2516.
Dale EJ,Vermeulen NA,Thomas AA,Barnes JC,M,Blackburn AK,Strutt NL,Sarjeant AA,Stern CL,Denmark SE,Stoddart JF,“ExCage,”J.Am.Chem.Soc.136:10669-82(2014).
Hafezi N,Holcroft JM,Hartlieb KJ,Dale EJ,Vermeulen NA,Stern CL,Sarjeant AA,Stoddart JF,“Modulating the binding of polycyclic aromatic hydrocarbons inside a hexacationic cage by anion-πinteractions,”Angew.Chem.Int.Ed.Engl.54:456-61(2015).
本文中引用的所有专利、专利申请、专利申请公报和其他出版物均整体上一并引入作为参考,就如其整体被提出一样。
以上用目前被认为最实用和优选的实例描述了本发明。但是,目前解释说明本发明的方式不意味着其限于所公开的实例。相应地,本领域技术人员将会意识到本发明旨在包含所有立足于发明主旨和权利要求范围的改变和替代方式。
- 还没有人留言评论。精彩留言会获得点赞!