一种乌苯美司δ晶型及其制备方法与流程
本申请涉及药物晶型领域,具体涉及一种乌苯美司δ晶型及其制备方法。
背景技术:
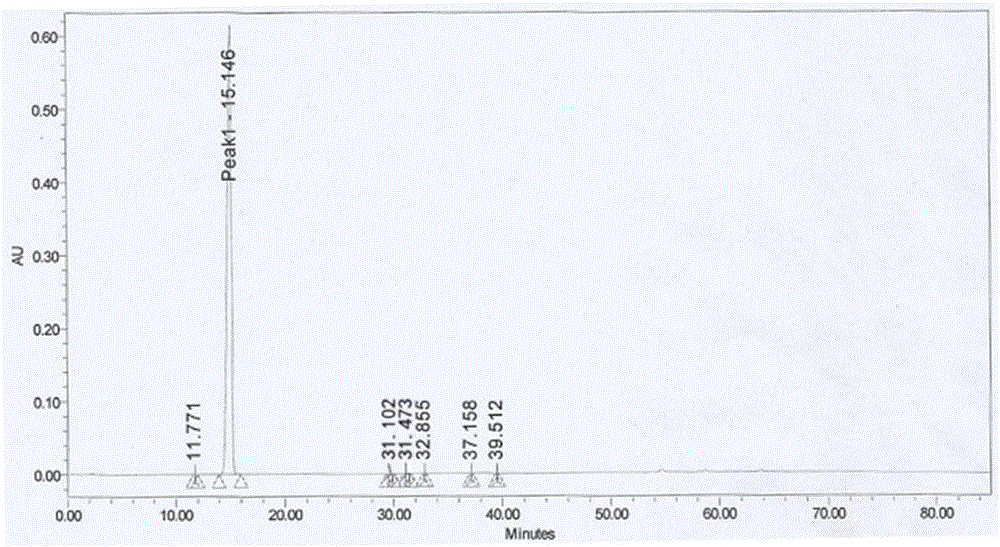
:乌苯美司(Ubenimex),其化学名称为:N-[(2S,3R)-3-氨基-2-羟基-4-苯基丁酰]-L-亮氨酸;分子式:C16H24N2O4;分子量:308.37;其结构式如下:乌苯美司是1976年由日本学者梅泽滨夫从橄榄网状链霉菌的培养液中分离而得的二肽化合物,可竞争性的抑制氨肽酶B及亮氨酸肽酶和半胱天冬酶等,诱导肿瘤细胞凋亡和促进宿主免疫功能。可配合化疗、放疗及联合应用于白血病、多发性骨髓瘤、骨髓增生异常综合征及造血干细胞移植后,以及其它实体瘤患者。US4281180公开了乌苯美司的α、β晶型的制备工艺,其中,在乌苯美司中间体用钯炭氢化反应时,容易产生过度氢化杂质。在后续加盐酸溶解,然后滴加氨水调pH值析晶的过程中,该杂质无法被有效的除掉。US4786754公开了乌苯美司γ晶型,并指出β晶型在空气中通过吸收水分会变成二水合物α晶型,而α晶型在148°下干燥可以得到β晶型,最终α、β晶型均会转变为稳定型γ晶型。因此α、β晶型存在不稳定导致产品很难控制的问题。同时,由于γ晶型是通过α、β制备而来,因此,同样也存在由于过度氢化导致杂质难以有效去除的问题。中国专利CN103910648A提供了一种盐酸乌苯美司的晶型制备方法:将盐酸乌苯美司加入到乙酸异丙酯与二甲基甲酰胺的混合溶剂中微热溶解,搅拌1-5h,静置使溶剂挥发后得到晶型。这种方法挥发时间长、只适合于小剂量晶型制备,无法满足大批量晶型制备要求。众所周知,对于药物的多晶型而言,不同的晶型可以具有不同的化学和物理特性,包括化学稳定性、溶解度、光学和机械性质等,这些性质可以直接影响原料药和制剂的处理和生产工艺过程,并且会影响到制剂的稳定性、溶解度和生物利用度。因此,晶型的研究对于药物制剂的质量、安全性和有效性具有重要意义。技术实现要素:本申请在不断地研究过程中,通过试验摸索,意外的获得了一种乌苯美司的δ晶型。该晶型显示了良好的稳定性,同时具有更好的流动性,更适合工业化大生产所需。本申请提供了乌苯美司的一种新的δ晶型,使用Cu-ka射线进行X射线粉末测定,其图谱具有下表所示的衍射角、晶面间距和相对强度:2θ衍射角的误差为±0.20。该晶型的DSC图谱在244.43℃℃有特征吸收峰,TGA显示该结晶样品加热至110℃时失重0.05%。对该结晶样品用KF法进行水分测定得出其含水量为0.02%。该晶型为新晶型,定义为乌苯美司δ晶型。更进一步的,该晶型具有基本如附图1所示的X射线粉末衍射图谱。本申请还公开了乌苯美司δ晶型的制备方法,具体包含以下步骤:(1)将乌苯美司溶解于乙醇和纯化水的混合溶剂中,乙醇和纯化水的用量比例为(1:4)~(3:1),乌苯美司与混合溶剂的用量比例为1g:(20~60ml),溶解温度为40~60℃;(2)向步骤(1)所得溶液加入活性炭,活性炭与乌苯美司的用量比例为:(0.01~0.1):1,脱色温度40~70℃,脱色时间为0.5h~4h;(3)将步骤(2)所得溶液进行抽滤;(4)在180r/min~200r/min转速搅拌下将步骤(3)所得滤液降温析晶,析晶时间为2h~6h,析晶温度为5~25℃,;(5)将步骤(4)所得溶液进行抽滤;(6)将步骤(5)所得滤饼置入真空烘箱里干燥4h~24h,温度条件为50~80℃。其中,步骤(1)中,乙醇和纯化水的用量比例优选为(1:2)~(2:1),乌苯美司与混合溶剂的用量比例优选为1g:(30ml~40ml),溶解温度优选为50~60℃。其中,步骤(2)中,活性炭与乌苯美司的用量比例优选为:(0.02~0.05):1,脱色温度优选为50~60℃,脱色时间为优选为0.5h~2h。其中,步骤(4)中,搅拌转速优选为180r/min~190r/min,析晶时间优选为3h~4h,析晶温度优选为10~15℃;其中,步骤(6)中,所述干燥时间优选为8h~16h,所述温度条件优选为70~80℃。采用本申请制备的乌苯美司δ晶型具有以下优点:1、通过溶解性、影响因素实验显示,本申请制备的δ晶型具有更好的溶解性和稳定性;通过粒度、休止角测试实验显示,本申请的晶型粒度较γ晶型更大,休止角较γ晶型更小,流动性更好。众所周知,乌苯美司是难溶性药物,粒径的大小,流动性的优劣会对后期制剂产品的溶出以及最终质量产生较大影响,因此,本申请δ晶型相比现有晶型更具有制剂优势。2、本申请晶型制备反应条件温和、操作简单,所用溶剂仅为乙醇和水,利于环保,回收利用方便,降低了试剂成本和能耗,而现有γ晶型的制备是先制备乌苯美司的α、β晶型,再通过α、β在有机溶剂中搅拌进行转化得到,所用溶剂包括甲乙酮、二乙酮、乙酸乙酯、二恶烷等,这些溶剂毒性大,且对环境污染严重。3、通过对δ晶型制备过程中的各关键参数控制,本申请制备方法能够显著提高δ晶型的收率,并得到单一晶型,工艺重现性好,纯度高(HPLC纯度>99.8%),使得实现工业化生产条件可控,有利于大规模产业化。附图说明图1实施例1乌苯美司δ晶型的XRPD图谱图2实施例1乌苯美司δ晶型的HPLC图谱图3对比例乌苯美司γ晶型的XRPD图谱图4实施例1乌苯美司δ晶型的粒度分布图图5对比例乌苯美司γ晶型的粒度分布图具体实施方式以下将结合实施例对本申请作进一步的详细描述,本申请的实施例仅用于说明本申请的技术方案,并非限制本申请的实质和范围。化合物的结构是通过核磁共振(1HNMR)来测定的。核磁共振(1HNMR)位移(δ)以百万分之一(ppm)为单位给出;核磁共振(1HNMR)的测定是用BrukerAVANCE-400核磁仪进行,测定溶剂为六氘代二甲基亚砜(CDCl3),内标为四甲基硅烷(TMS)。HPLC谱图的测定采用安捷伦Agilent1260DAD型液相色谱仪进行。本申请所述的X射线粉末衍射(XRPD)的测定是采用辽宁丹东浩元DX-2700X粉末衍射仪进行采集,具体参数如下表:本申请所述的差示扫描量热分析(DSC)和热重分析(TGA)测定数据分别在TAQ2000差示扫描量热仪和TAQ5000热重分析仪上采集,仪器参数如下表所示:DSCTGA样品盘铝盘,压盖铂金盘,敞开温度范围室温-300℃室温-350℃扫描速率10℃/分钟10℃/分钟保护气体氮气氮气本申请粒度测试所使用的仪器为激光粒度分析仪LS-C(III),由珠海欧美克仪器有限公司提供。本申请所述“室温”是指温度处于10~25℃之间。本申请中,乌苯美司HPLC纯度用以下方法进行:(1)以十八烷基硅烷键合硅胶为填充剂的色谱柱(5μm粒径,柱温25℃);(2)流动相A:稀释的0.1mol/L磷酸二氢钾溶液(13→20)和乙腈混合物(V/V=17/3);流动相B:乙腈和稀释的0.1mol/L的磷酸二氢钾溶液(13→20)混合物(V/V=2/1);流动相A和B的洗脱梯度见下表:(3)检测器:紫外吸收光度计(波长:220nm);(4)流速:调整流速,使得乌苯美司的保留时间约为14分钟;(5)运行时间:从溶剂峰开始计时,约为乌苯美司保留时间的5倍;(6)标准溶液:将30毫克乌苯美司溶解在10毫升的流动相A中,作为样品溶液。吸取2毫升样品溶液,加流动相A稀释到200毫升;(7)进样量:20μl,通过自动积分法测定样品溶液按峰面积计算样品中乌苯美司的纯度。具体实施方式:实施例1乌苯美司δ晶型的制备将10g乌苯美司溶解于200ml乙醇和200ml纯化水的混合溶剂中,溶解温度为60℃,溶解完全后加入1.0g活性炭脱色1h,脱色温度为60℃;负压抽滤,所得滤液在180r/min转速搅拌下降温析晶,析晶温度为10℃,析晶时间为3h;负压抽滤,滤饼置入70℃真空烘箱里干燥8h,得白色固体粉末9.25g,收率92.5%,HPLC纯度为99.89%,总杂0.11%,最大单杂0.06%。其核磁共振结果为:1HNMR(400MHz,DMSO):δ7.30(s,5H),3.99(s,1H),3.89(s,1H),3.57(s,1H),2.92(m,2H),1.90(s,1H),1.60(s,2H),0.85(d,J=4.7Hz,6H)13CNMR(101MHz,DMSO):δ175.07(s,1C),171.48(s,1C),137.13(s,1C),129.50(s,2C),128.48(s,2C),126.64(s,1C),68.48(s,1C),55.20(s,1C),52.77(s,1C),35.43(s,1C),24.71(s,1C),23.23(s,1C),21.32(d,J=14.7Hz,2C)该结晶样品的DSC在244.43℃有特征吸收峰,TGA显示该结晶样品加热至110℃时失重0.05%。对该结晶样品用KF法进行水分测定得出其含水量为0.02%。根据以上结果得知,该晶型为新晶型,定义为乌苯美司δ晶型。使用Cu-ka射线进行X射线粉末测定,其图谱具有下表所示衍射角、晶面间距和相对强度:2θ衍射角的误差为±0.20。更进一步的,实施例1的乌苯美司δ晶型具有基本如附图1所示的X射线粉末衍射图谱。实施例2:乌苯美司δ晶型的制备将乌苯美司16g溶解于乙醇320ml和纯化水160ml的混合溶剂中,溶解温度为60℃,溶解完全后加活性炭1.28g脱色2h,脱色温度为50℃;负压抽滤,滤液在200r/min转速搅拌下降温析晶,析晶温度为15℃,析晶时间为4h;负压抽滤,滤饼置入75℃真空烘箱里干燥16h,得白色固体粉末14.6g,收率91.25%。HPLC纯度为99.88%,总杂0.12%,最大单杂0.07%。其核磁数据、DSC数据、粉末衍射数据与实施例1数据基本保持一致。实施例3:乌苯美司δ晶型的制备将乌苯美司10g溶解于乙醇180ml和纯化水320ml的混合溶剂中,溶解温度为55℃,溶解完全后加活性炭1.0g脱色2h,脱色温度为55℃;负压抽滤,滤液在180r/min转速搅拌下降温析晶,析晶温度为5℃,析晶时间为3h;负压抽滤,滤饼置入80℃真空烘箱里干燥4h,得白色固体粉末8.96g,收率89.6%。HPLC纯度为99.90%,总杂0.10%,最大单杂0.05%。其核磁数据、DSC数据、粉末衍射数据与实施例1数据基本保持一致。实施例4:乌苯美司δ晶型的制备将乌苯美司50g溶解于乙醇800ml和纯化水1600ml的混合溶剂中,溶解温度为50℃,溶解完全后加活性炭0.5g脱色0.5h脱色温度为70℃;负压抽滤,滤液在200r/min转速搅拌下降温析晶,析晶温度为8℃,析晶时间为5h;负压抽滤,滤饼置入80℃真空烘箱里干燥20h,得白色固体粉末44.35g,收率88.7%。HPLC纯度为99.85%,总杂0.14%,最大单杂0.08%。其核磁数据、DSC数据、粉末衍射数据与实施例1数据基本保持一致。实施例5:乌苯美司δ晶型的制备将乌苯美司8g溶解于乙醇125ml和纯化水180ml的混合溶剂中,溶解温度为60℃,溶解完全后加活性炭0.8g脱色0.5h,脱色温度为60℃;负压抽滤,滤液在200r/min转速搅拌下降温析晶,析晶温度为12℃,析晶时间为2h;负压抽滤,滤饼置入60℃真空烘箱里干燥10h,得白色固体粉末7.46g,收率93.25%。HPLC纯度为99.91%,总杂0.10%,最大单杂0.05%。其核磁数据、DSC数据、粉末衍射数据与实施例1数据基本保持一致。实施例6乌苯美司δ晶型的制备将乌苯美司10g溶解于乙醇100ml和纯化水100ml的混合溶剂中,溶解温度为60℃,溶解完全后加活性炭0.5g脱色4h,脱色温度为40℃;负压抽滤,滤液在,190r/min转速搅拌下降温析晶,析晶温度为25℃,析晶时间为6h;负压抽滤,滤饼置入50℃真空烘箱里干燥24h,得白色固体粉末9.53g,收率95.3%。HPLC纯度为99.87%,总杂0.13%,最大单杂0.04%。其核磁数据、DSC数据、粉末衍射数据与实施例1数据基本保持一致。实施例7乌苯美司δ晶型的制备将乌苯美司10g溶解于乙醇100ml和纯化水300ml的混合溶剂中,溶解温度为60℃,溶解完全后加活性炭0.2g脱色4h,脱色温度为40℃;负压抽滤,滤液在190r/min转速搅拌下降温析晶,析晶温度为25℃,析晶时间为6h;负压抽滤,滤饼置入50℃真空烘箱里干燥24h,得白色固体粉末9.67g,收率96.7%。HPLC纯度为99.86%,总杂0.14%,最大单杂0.06%。其核磁数据、DSC数据、粉末衍射数据与实施例1数据基本保持一致。对比例样品:乌苯美司γ晶型的制备按照US4786754专利实施例1公开的内容,将通过等电点沉淀法获得的乌苯美司α晶型100g,放入150℃烘箱里干燥3h,得到经XPRD验证的乌苯美司γ晶型91g。其粉末衍射数据XPRD测试结果见下表和附图3。试验例1溶解性考察试验为了考察本申请δ晶型与γ晶型在溶解上的区别,本申请分别在25℃、37℃条件下在pH=1.0(0.1N)的盐酸和pH=4.5的乙酸-乙酸钠缓冲溶液中,测定平衡溶解度(饱和溶液),结果如下表1所示:表1溶解性试验溶解性试验结果表明,上述各种条件下,与对比γ晶型相比,本申请乌苯美司δ晶型溶解性均更好。试验例2化学稳定性考察试验将实施例1~5制备的乌苯美司δ晶型和对比例制备的乌苯美司γ晶型样品分别放入洁净的培养皿中敞口平摊放置,考察在高温(60℃)、高湿(25℃、RH90%±5%)、强光(1.2×106Lux)条件下样品的稳定性,考察取样时间为5天和10天,放置10天。分别在5天和10天取样测定,HPLC纯度检测结果见下表2:表2稳定性试验纯度测定结果稳定性考察试验结果表明,在光照、高温、高湿三种条件下,本申请乌苯美司δ晶型纯度基本没有发生变化,而对比例样品的数据相对变化明显,即说明本申请的晶型的稳定性较对比γ晶型更优异。试验例3粒度测试和流动性评价为了比较本申请实施例1制备的δ晶型样品和对比例γ晶型样品在粒度分布和流动性的差异,申请人分别按照激光散色衍射法和注入法测定粒度分布和休止角,结果如表3所示:表3粒度、休止角测试结果从表3可以看出,本申请实施例1制备的δ晶型77.88%粒径为32-65μm;而对比例γ晶型占比86.99%的粒径为3-32μm,即本申请δ晶型粒度大于γ晶型。对于同一种物料,粒径愈小,颗粒间相互粘附力越大、流动性越差。本申请δ晶型休止角为为38.66°,对比例样品休止角为50.43°。同一物质,休止角越小,流动性越好。这两个结果均证明,δ晶型与γ晶型相比具有更好的流动性。众所周知,乌苯美司是难溶性药物,对于难溶性药物而言,原料药的粒度、休止角会影响到后期制剂产品压片、溶出效果等,最终会影响到产品质量。流动性越差,后期制剂过程中往往需要通过通过加入助流剂等来改善流动性。而对于药品而言,在满足药品质量符合药品注册要求的前提下,辅料的用量应越少越好。因此,相比于对比例γ晶型而言,本申请的δ晶型粒度更大,休止角更小,在粉体流动性方面的优势更明显,后期制剂过程中部不需要刻意改善流动性,更具有制剂优势,更适合用于工业化大生产。综上所述,本申请提供了一种乌苯美司新的δ晶型,该晶型具有优异的稳定性,更好地溶解性;通过粒度、休止角测试实验显示,本申请的晶型粒度较γ晶型更大,休止角较γ晶型更小,即相对于γ晶型,本申请制备的晶型具有更好的流动性和制剂优势。本申请制备反应条件温和、操作简单,工艺重现性好,纯度高所用溶剂仅为乙醇和水,利于环保,回收利用方便,降低了试剂成本和能耗,与现有制备工艺需使用毒性大的有机溶剂相比,更环保,更适合工业化大生产。对于本领域的普通技术人员而言明显的是,在不偏离本申请精神或者范围的情况下,可对本申请化合物、组合物以及其制备方法进行的多种修饰和变化,因此,本申请的保护范围涵盖了对本申请进行的各种修饰和变化,只要所述修饰或变化处于权利要求和其等同实施方式所涵盖的范围内。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1