一种高光学纯度联苯基丙氨酸及其衍生物的制备方法和应用与流程
本发明属于药物合成领域,具体涉及一种联苯基丙氨酸及其衍生物的制备方法和应用。
背景技术:
LCZ696是由诺华(Novartis)公司开发的一种新型降压药物,该药结合了缬沙坦和实验性药物Sacubitril(AHU-377)等两种组份,其中缬沙坦可改善血管舒张,刺激身体排泄钠和水,而Sacubitril可阻断威胁降低血压的2种多肽的作用机制,因而LCZ696被称为血管紧张素II受体与脑啡肽酶的双重抑制剂,业界普遍认为LCZ696将会带来传统心衰治疗方案的革新。
Sacubitril的化学名为:4-(((2S,4R)-1-(1,1,-联苯-4-基)-5-乙氧基-4-甲基-5-氧代戊烷-2-基)氨基)-4-氧代丁酸(I),其结构式为:
联苯基丙氨酸衍生物是Sacubitril的一个关键中间体,特别是具有光学纯度的联苯基丙氨酸衍生物,且该中间体在中性内中性肽链内切酶(NEP)抑制剂中也具有广泛的应用如文献US4722810、US5223516、US4610816、WO92/14706、US4929641、US5217996、US5273990、US5294632、US5250522、WO93/09101、EP00590442、WO93/10773、WO2008/031567中报道。
现有技术中报道具有光学纯度的联苯基丙氨酸衍生物的制备方法主要有以下几种:
(1)US5217996,Journal of Medicinal Chemistry 1995,38(10),1689-1700报道将D-Boc-酪氨酸甲酯转化为三氟甲磺酸酯,再将所述三氟甲磺酸酯与苯硼酸进行Suzuki反应,再将所得酯水解,合成方法如下:
该方法用于制备D-Boc-酪氨酸甲酯的原料为非天然的D-酪氨酸,该化合物价格昂贵,此外该方法需要三氟甲磺酸酐以活化酚羟基从而进行芳基偶联反应,三氟甲磺酸酐试剂及偶联反应的钯催化剂价格也很贵,因此该方法开发成本很高,工业实用性很差。
(2)JP2003261522A,Chirality,1996,8(2),173-188报道通过不对称加氢合成旋光Boc-联苯基丙氨酸的方法,该方法需要使用不对称加氢催化剂,该类不对称加氢催化剂的价格也非常昂贵,且手性纯度不能得到保证,因此该方法开发成本同样很高,工业实用性同样很差。
因此有必要开发一条适合工业制备光学纯联苯基丙氨酸及其衍生物的方法。
技术实现要素:
为了解决现有技术存在的缺陷,发明人在深入研究以后开发出一种制备高光学纯度联苯基丙氨酸及其衍生物的方法,以及其作为关键中间体用于制备Sacubitril的应用。
本发明一方面提供了一种高光学纯度式(Ⅰ)化合物的制备方法,包括如下步骤:在第一溶剂中,式(Ⅱ)化合物在酸性拆分试剂的存在下反应生成酸式盐,然后在第二溶剂中,控制反应液pH值为6.5-12.5反应生成得到式(Ⅰ)化合物,合成路线如下:
其中,R1选自氢、氘、C1-8烷基、C2-8链烯基、C2-8链炔基、C3-8环烷基、3-8元杂环基、C5-10芳基或5-10元杂芳基,任选进一步被一个或多个选自卤素、羟基、巯基、氰基、硝基、叠氮基、甲基、乙基、异丙基、三氟甲基、乙烯基、烯丙基、乙炔基、环丙基、四氢呋喃基、吗啉基、苯基、甲氧基、乙氧基、异丙氧基、苄氧基、磺酰基、甲磺酰基、异丙磺酰基、对甲苯磺酰基、甲氧羰基、乙氧羰基、异丙氧羰基、乙酰基或乙酰氧基取代基所取代,所述杂原子选自O或S;
R2、R3各自独立的选自氢、氘、卤素、羟基、巯基、氰基、硝基、叠氮基、C1-8烷基、C2-8链烯基、C2-8链炔基、卤取代C1-8烷基、C3-8环烷基、3-8元杂环基、C1-8烷氧基、C3-8环烷氧基、3-8元杂环基氧基、3-8元杂环基硫基、C5-10芳基、C5-10芳基氧基、C5-10芳基硫基、5-10元杂芳基、5-10元杂芳基氧基、5-10元杂芳基硫基、C1-8烷基磺酰基、C1-8烷氧羰基、C1-8烷酰基或C1-8烷酰氧基;
m、n各自独立的选自0、1、2、3、4、5。
优选地,所述的制备方法中R1选自氢、氘、C1-8烷基、C2-8链烯基、C2-8链炔基、C3-8环烷基、3-8元杂环基、C5-10芳基或5-10元杂芳基,任选进一步被一个或多个选自卤素、羟基、甲基、乙基、异丙基、三氟甲基、乙烯基、烯丙基、乙炔基、环丙基、苯基、甲氧基、乙氧基、异丙氧基、苄氧基、甲氧羰基、乙氧羰基、异丙氧羰基、乙酰基或乙酰氧基取代基所取代,所述杂原子选自O或S;R2、R3、m、n如式(Ⅰ)化合物所定义。
进一步优选地,所述的制备方法中R1选自氢、氘、C1-8烷基、C2-8链烯基、C2-8链炔基、C3-8环烷基或C5-10芳基,任选进一步被一个或多个选自卤素、甲基、乙基、异丙基、环丙基、苯基、甲氧基、乙氧基、异丙氧基、苄氧基、甲氧羰基、乙氧羰基、异丙氧羰基、乙酰基或乙酰氧基取代基所取代;R2、R3各自独立的选自氢、氘、卤素、羟基、氰基、硝基、叠氮基、C1-8烷基、C2-8链烯基、C2-8链炔基、卤取代C1-8烷基、C3-8环烷基、C1-8烷氧基、C3-8环烷氧基、C5-10芳基、C5-10芳基氧基、C1-8烷基磺酰基、C1-8烷氧羰基、C1-8烷酰基或C1-8烷酰氧基;m、n如式(Ⅰ)化合物所定义。
更进一步优选地,所述的制备方法中R1选自C1-8烷基、C2-8链烯基、C2-8链炔基、C3-8环烷基或苯基,任选进一步被一个或多个选自卤素、环丙基、苯基、甲氧基、乙氧基、异丙氧基或苄氧基的取代基所取代;R2、R3各自独立的选自氢、氘、卤素、羟基、C1-8烷基、卤取代C1-8烷基、C3-8环烷基、C1-8烷氧基、C3-8环烷氧基、C5-10芳基、C5-10芳基氧基、C1-8烷氧羰基、C1-8烷酰基或C1-8烷酰氧基;m、n如式(Ⅰ)化合物所定义。
最优选地,所述的制备方法中R1选自C1-8烷基、C3-8环烷基或苯基,任选进一步被一个或多个选自卤素、环丙基、苯基、甲氧基、乙氧基、异丙氧基或苄氧基的取代基所取代;R2、R3各自独立的选自氢、氘、卤素、C1-8烷基、卤取代C1-8烷基或C3-8环烷基;m、n各自独立的选自0、1或2。
在一些实施方案中,所述的制备方法中制备得到的高光学纯度式(Ⅰ)化合物的ee值大于98.0%;优选大于99.0%。
在一些实施方案中,所述的制备方法中反应生成其酸式盐后控制反应液pH值为7.5-10.5,优选控制反应液pH值为8.5-10.0。
在一些实施方案中,所述的制备方法中所采用的酸性拆分试剂选自D-天冬氨酸、L-天冬氨酸、D-焦谷氨酸、L-焦谷氨酸、D-(-)-奎宁酸、D-(-)-酒石酸、L-(+)-酒石酸、(D)-(-)-扁桃酸、(L)-(+)-扁桃酸、(R)-(-)-1,1'-联萘酚磷酸酯、(S)-(-)-联萘酚磷酸酯、D(+)-10-樟脑磺酸、D(+)-樟脑酸、L(-)-樟脑磺酸、(+)-二乙酰基-L-酒石酸酐、(-)-二对甲苯酰-L-酒石酸、L-谷氨酸、D(+)-苹果酸、L(-)-苹果酸、(S)-(-)-α-甲苄基异氰酸酯或其混合物;优选D-(-)-酒石酸、(D)-(-)-扁桃酸、L-(-)樟脑磺酸、D-(-)-奎宁酸或其混合物。
在一些实施方案中,所述的制备方法中式(Ⅱ)化合物与酸性拆分试剂的摩尔比为1.0:0.5~5.0;优选摩尔比为1.0:0.8~1.5;更优选摩尔比为1.0:0.9~1.2。
在一些实施方案中,所述制备方法中第一溶剂、第二溶剂各自独立选自二氯甲烷、乙酸乙酯、乙酸异丙酯、乙酸正丁酯、甲苯、环己烷、甲基叔丁基醚、异丙醚、丙酮、甲醇、乙醇、异丙醇、正丙醇、正丁醇、叔丁醇、四氢呋喃、二氧六环、乙腈、二甲亚砜、N、N-二甲基甲酰胺或其混合物;优选二氯甲烷、乙酸乙酯、乙酸异丙酯、丙酮或异丙醇。
优选地,所述制备方法所采用的第一溶剂和/或第二溶剂进一步包括水。
在一些实施方案中,所述的制备方法中控制反应液pH值所用试剂选自有机碱、无机碱或其混合物,所述有机碱选自氨水、甲胺、乙胺、乙醇胺、乙二胺、三甲胺、三乙胺、丙胺、异丙胺、1,3-丙二胺、三乙醇胺、二异丙基乙胺、吡啶、哌啶、吗啉或其混合物,优选氨水或三乙胺;所述无机碱选自碳酸钾、碳酸氢钠、碳酸氢钾、碳酸钠、氢氧化钾、氢氧化锂、醋酸钠或其混合物,优选碳酸氢钠、氢氧化钠、或碳酸氢钾。
在一些实施方案中,所述的制备方法中控制反应液pH值所用试剂可以直接加入或用合适溶剂溶解后加入;加入方式可以分批加入或滴加加入;优选的,所述合适溶剂选自前述的第一溶剂或第二溶剂。
在一些实施方案中,所述制备方法反应生成酸式盐后,进一步包括精制的步骤。
优选地,所述精制的步骤具体为:将酸式盐加入第三溶剂中,加热溶解,缓慢冷却至0-25℃,过滤,滤饼干燥。
进一步优选地,所述精制的步骤中第三溶剂选自丙酮、甲醇、乙醇、异丙醇、正丙醇、正丁醇、叔丁醇、四氢呋喃、二氧六环、乙腈、二甲亚砜、N、N-二甲基甲酰胺或其混合物,优选丙酮、异丙醇或四氢呋喃。
本发明另一方面提供了一种式(Ⅱ)化合物的制备方法,合成路线如下:
其中,X选自卤素,优选氯或溴;R1、R2、R3、m、n如式(Ⅰ)化合物所定义。
本发明再一方面提供了一种所述的制备方法在制备AHU-377或其类似物中的应用,还包括如下合成步骤:
其中,Pg为氨基保护基,优选叔丁氧羰基、烯丙基羰基、笏甲氧羰基、甲氧羰基、乙氧羰基、三甲基硅乙氧羰基或苄氧羰基;R1、R2、R3、m、n如式(Ⅰ)化合物所定义。
与现有技术相比,本发明具有以下优势:
1、本发明采用酸性拆分试剂进行拆分,原料简单易得,价格便宜,有利于降低产品的开发成本。
2、本发明拆分后产物的光学纯度高,解决了现有技术中难以精制得到高光学纯度最终产物的问题。
3、本发明操作简单,安全性高,废水用量少,能量消耗少,可以有效解决药物开发中的EHS(环境、健康、安全)问题。
附图说明
图1为实施例1产物(R)-2-氨基-3-联苯基丙酸乙酯手性HPLC图谱;
图2为实施例4产物AHU-377手性HPLC图谱。
具体实施方式
详细说明:除非有相反陈述,下列用在说明书和权利要求书中的术语具有下述含义。
“C1-8烷基”指包括1至8个碳原子的直链烷基和含支链烷基,烷基指饱和的脂族烃基团,例如甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、仲丁基、正戊基、1,1-二甲基丙基、1,2-二甲基丙基、2,2-二甲基丙基、1-乙基丙基、2-甲基丁基、3-甲基丁基、正己基、1-乙基-2-甲基丙基、1,1,2-三甲基丙基、1,1-二甲基丁基、1,2-二甲基丁基、2,2-二甲基丁基、1,3-二甲基丁基、2-乙基丁基、2-甲基戊基、3-甲基戊基、4-甲基戊基、2,3-二甲基丁基、正庚基、2-甲基己基、3-甲基己基、4-甲基己基、5-甲基己基、2,3-二甲基戊基、2,4-二甲基戊基、2,2-二甲基戊基、3,3-二甲基戊基、2-乙基戊基、3-乙基戊基、正辛基、2,3-二甲基己基、2,4-二甲基己基、2,5-二甲基己基、2,2-二甲基己基、3,3-二甲基己基、4,4-二甲基己基、2-乙基己基、3-乙基己基、4-乙基己基、2-甲基-2-乙基戊基、2-甲基-3-乙基戊基或其各种支链异构体等。
“环烷基”指饱和或部分不饱和单环或多环环状烃取代基,“C3-8环烷基”指包括3至8个碳原子的环烷基,例如:
单环环烷基的非限制性实施例包含环丙基、环丁基、环戊基、环戊烯基、环己基、环己烯基、环己二烯基、环庚基、环庚三烯基、环辛基等。
多环环烷基包括螺环、稠环和桥环的环烷基。“螺环烷基”指单环之间共用一个碳原子(称螺原子)的多环基团,这些可以含有一个或多个双键,但没有一个环具有完全共轭的π电子系统。根据环与环之间共用螺原子的数目将螺环烷基分为单螺环烷基、双螺环烷基基或多螺环烷基,螺环烷基的非限制性实施例包含:
“稠环烷基”指系统中的每个环与体系中的其他环共享毗邻的一对碳原子的全碳多环基团,其中一个或多个环可以含有一个或多个双键,但没有一个环具有完全共轭的π电子系统。根据组成环的数目可以分为双环、三环、四环或多环稠环烷基,稠环烷基的非限制性实施例包含:
“桥环烷基”指任意两个环共用两个不直接连接的碳原子的全碳多环基团,这些可以含有一个或多个双键,但没有一个环具有完全共轭的π电子系统。根据组成环的数目可以分为双环、三环、四环或多环桥环烷基,桥环烷基的非限制性实施例包含:
所述环烷基环可以稠合于芳基、杂芳基或杂环烷基环上,其中与母体结构连接在一起的环为环烷基,非限制性实施例包括茚满基、四氢萘基、苯并环庚烷基等。
“烷氧基”指-O-(烷基),其中烷基的定义如上所述。“C1-8烷氧基”指含1-8个碳的烷基氧基,非限制性实施例包含甲氧基、乙氧基、丙氧基、丁氧基等。
“环烷氧基”指和-O-(未取代的环烷基),其中环烷基的定义如上所述。“C3-8环烷氧基”指含3-8个碳的环烷基氧基,非限制性实施例包含环丙氧基、环丁氧基、环戊氧基、环己氧基等。
“卤取代的C1-8烷基”指烷基上的氢任选的被氟、氯、溴、碘原子取代的1-8个碳烷基基团,例如二氟甲基、二氯甲基、二溴甲基、三氟甲基、三氯甲基、三溴甲基等。
下面结合实施例对本发明做进一步详细、完整地说明,但决非限制本发明,本发明也并非仅局限于实施例的内容。
本发明的化合物HPLC的测定使用安捷伦1260高效液相色谱仪,手性HPLC方法为:柱型号:CHIRALPAK AD-H,0.46cm×25cm×5μm;流动相:己烷/异丙醇=80/20(v/v);流速:1.0ml/min;波长:254nm。
薄层层析硅胶板使用烟台黄海HSGF254或青岛GF254硅胶板,TLC采用的规格是0.15mm~0.20mm,薄层层析分离纯化产品采用的规格是0.4mm~0.5mm。柱层析一般使用烟台黄海硅胶200~300目硅胶为载体。
本发明实施例中的起始原料是已知的并且可以在市场上买到,或者可以采用或按照本领域已知的方法来合成。
在无特殊说明的情况下,本发明的所有反应均在连续的磁力搅拌下,在干燥氮气或氩气氛下进行,溶剂为干燥溶剂。
实施例1
步骤1:二苯甲酮甘氨酸亚胺乙酯的制备
向反应瓶中加入甘氨酸乙酯139.6g,二苯甲酮400g,对甲苯磺酸(PTS)9.5g,甲苯400ml,升温至回流,慢慢往反应体系中滴加二异丙基乙胺。二异丙基乙胺滴加完后,继续反应2小时。反应液冷却至室温,加入水300ml,甲苯200ml,搅拌,静置分层,甲苯层用盐水洗涤1次。将甲苯层进行浓缩至干得到液体二苯甲酮甘氨酸亚胺乙酯490g,产率93%。
步骤2:N-二苯基亚甲基-2-(4-联苯甲基)甘氨酸乙酯的制备
向反应瓶中加入步骤1中合成得到的二苯甲酮甘氨酸亚胺乙酯490g,再加入碳酸钾150g,乙腈1000ml,4-氯甲基联苯202g。升温至回流,回流反应5h,液相色谱监控反应完成。反应液冷却至室温,过滤,浓缩至干得到油状物650g,直接投入下步反应。
步骤3:2-氨基-3-联苯基丙酸乙酯的制备
在步骤2中合成得到的油状物中加入甲苯2000ml,水200ml,浓盐酸200ml,室温搅拌,慢慢析出固体,搅拌5h,室温抽滤,得到类白色固体。
在上述类白色固体中加入水200ml,二氯甲烷500ml,搅拌,往其中慢慢滴加氨水,调内液pH=10,分层,水层用二氯甲烷提取一次。合并二氯甲烷,饱和食盐水洗涤1次,硫酸钠干燥,过滤,浓缩至干得到油状物182.6g。
步骤4:(R)-2-氨基-3-联苯基丙酸乙酯-D-(-)-酒石酸盐的制备
向反应瓶中加入步骤3中合成得到的2-氨基-3-联苯基丙酸乙酯20g,再加入丙酮120g。将D-(-)-酒石酸11.5g溶于水11g和丙酮80g,室温下将酒石酸溶液慢慢滴加至反应瓶中,滴加过程中内温升至30℃,滴毕保持内温28~32℃搅拌反应20小时,将反应液冷却至10℃左右搅拌1小时,抽滤,干燥后得到白色固体。
将白色固体中加入四氢呋喃200g,升温至回流,冷却至室温,搅拌1小时,抽滤,干燥后得到白色固体12.7g,mp:177.1-177.5℃。
步骤5:(R)-2-氨基-3-联苯基丙酸乙酯的制备
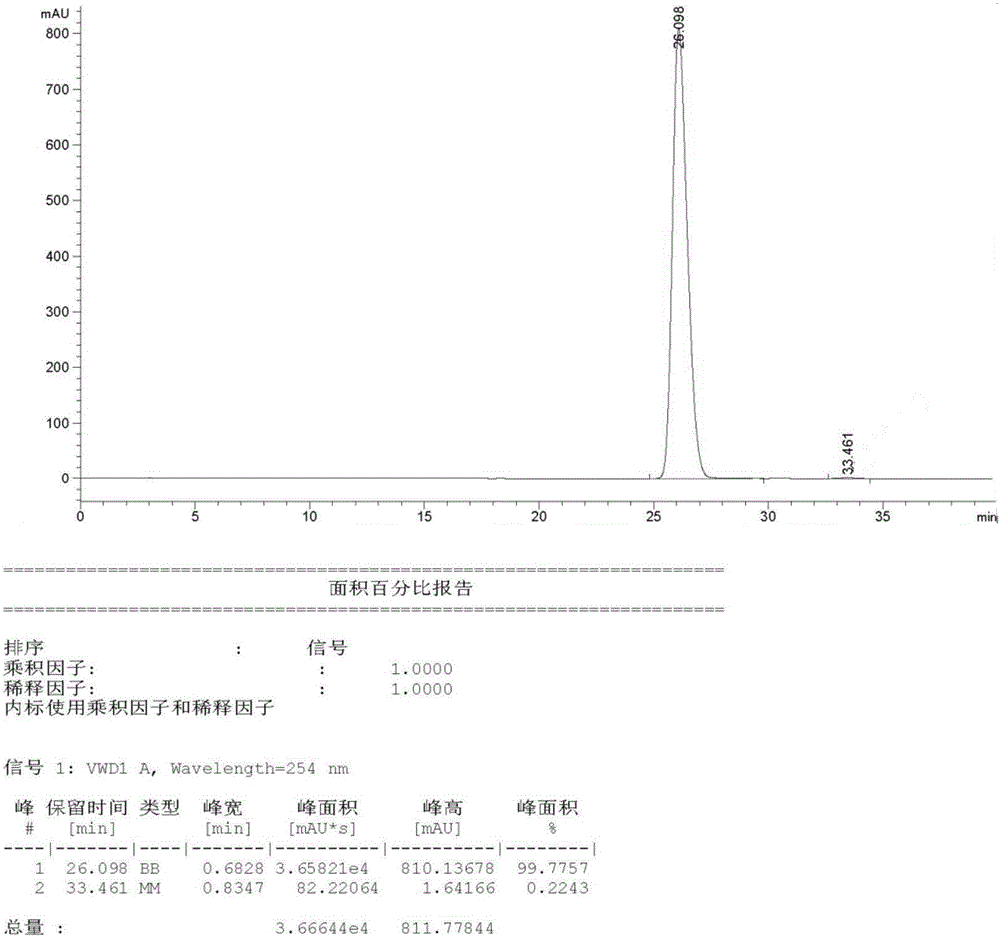
在步骤4中合成得到的(R)-2-氨基-3-联苯基丙酸乙酯-D-(-)-酒石酸盐中加入二氯甲烷250mL和水50mL,冰水冷却,搅拌,往体系中滴加氨水将反应液pH值调整至10.0-10.5,继续搅拌30分钟,分层,有机层用饱和食盐水洗涤2次,硫酸钠干燥,浓缩至干得到无色液体7.5g,ee值为99.52%(手性HPLC分析),手性HPLC图谱见附图1。
实施例2
步骤1:制备(R)-2-氨基-3-联苯基丙酸乙酯-D-(-)-扁桃酸盐
向反应瓶中加入2-氨基-3-联苯基丙酸乙酯20g,再加入异丙醇120g。将D-(-)-扁桃酸13.8g溶于水15g和异丙醇80g,室温下将D-(-)-扁桃酸溶液慢慢滴加至反应瓶中,滴加过程中内温升至30℃,滴毕保持内温在28~32℃搅拌20小时,将反应液冷却至10℃左右继续搅拌1小时,抽滤,干燥后得到白色固体。
往白色固体中加入异丙醇200g,升温至回流,冷却至室温,继续搅拌1小时,抽滤,干燥后得到白色固体12.6g。
步骤2:(R)-2-氨基-3-联苯基丙酸乙酯的制备
在步骤1合成的(R)-2-氨基-3-联苯基丙酸乙酯-D-(-)-扁桃酸盐中加入乙酸异丙酯220mL和水80mL,冰水冷却,搅拌下滴加三乙胺将反应液pH值调整至9.0-9.5,继续搅拌30分钟,分液,有机层用饱和食盐水洗涤2次,硫酸钠干燥,浓缩至干得到无色液体7.2g,ee值大于99.0%(手性HPLC分析),手性HPLC图谱基本与附图1一致。
实施例3
向反应瓶中加入2-氨基-3-联苯基丙酸乙酯20g,再加入乙酸乙酯150g。将L-(-)樟脑磺酸16.4g溶于水50g和丙酮50g,室温下将L-(-)樟脑磺酸溶液慢慢滴加至反应瓶中,滴加过程中内温升至30℃,滴毕保持内温在28~32℃继续搅拌反应20小时,将反应液冷却至10℃左右继续搅拌1小时,抽滤,丙酮洗涤滤饼,干燥,得到白色固体。
上述固体中加入二氯甲烷200mL和水50mL,冰水冷却,搅拌,往体系中滴加饱和碳酸氢钠水溶液将反应液pH值调整至7.5-8.5,继续搅拌60分钟,分液,有机层用饱和食盐水洗涤2次,硫酸钠干燥,浓缩至干得到无色液体6.9g,ee值大于99.0%(手性HPLC分析),手性HPLC图谱基本与附图1一致。
实施例4
下列化合物是根据以上一般方法制备得到的,具体过程参见实施例1-3。
实施例5 AHU-377的制备
步骤1:化合物1的制备
将16.5g(R)-2-氨基-3-联苯基丙酸乙酯加入150mL乙腈中,再加入26.74g Boc酸酐,室温搅拌反应2h,HPLC显示反应完全,减压蒸去乙腈,在残留物中加入200mL乙酸乙酯和40mL10%碳酸钠水溶液,搅拌30min,水相用100mL乙酸乙酯萃取,合并有机相,有机相分别用25mL水和25mL饱和食盐水各洗涤1次,无水硫酸钠干燥,浓缩至干得35.4g化合物1。
步骤2:化合物2的制备
将35.4g化合物1、17.5g LiOH·H2O溶于350mL丙酮和100mL水中,室温搅拌2h,HPLC显示反应完全,旋去大部分丙酮,在残留物中加入约200mL 2N HCl,调节pH=2-3,分别用250mL乙酸乙酯萃取3次,合并有机相,用50mL食盐水洗涤1次,无水硫酸钠干燥,过滤,浓缩至干得23.0g化合物2。
步骤3:化合物3的制备
将23.0g化合物2、7.89g N,O-二甲基羟胺盐酸盐、8.18g三乙胺、13.66g HOBT和25.83g EDCI溶于250mL二氯甲烷中,室温搅拌过夜。HPLC显示反应完全,旋干溶剂,在残留物中加入300mL乙酸乙酯和100mL碳酸氢钠水溶液,搅拌溶清,分去水层,有机相依次用100mL 1N HCl和50mL食盐水各洗涤1次,无水硫酸钠干燥,浓缩至干得22.7g化合物3。
步骤4:化合物4的制备
将22.3g化合物3溶于500mL THF中,氮气保护,冷却至-10℃,加入2.23g LiAlH4,温度上升至0℃左右,有气泡放出。30min后HPLC显示反应完全,加入23g KHSO4/100mL水淬灭反应,室温下继续搅拌1h。过滤除去不溶物,蒸干大部分THF,加入500mL乙酸乙酯和200mL1N盐酸,搅拌15min,过滤除去不溶物,分去水层,有机相用50mL食盐水洗涤1次,无水硫酸钠干燥,浓缩至干得18.7g化合物4。
步骤5:化合物5的制备
将16.5g化合物4溶于250mL二氯甲烷,加入36.93g乙氧甲酰基亚乙基三苯基膦,搅拌溶清,室温搅拌过夜。HPLC显示反应完全,浓缩至干,在残留物中再加入乙酸乙酯32mL和石油醚256mL,回流溶清,冷却析晶,过滤,烘干得到21.6g化合物5。
步骤6:化合物6的制备
将21.5g化合物5溶于500mL乙醇中,加入6.0g 5%钯炭,氮气置换三次,氢气置换三次,保持氢气压力0.3MPa,室温搅拌过夜,HPLC显示原料反应完全,浓缩至干,过柱分离得15.1g化合物6。
步骤7:化合物AHU-377的制备
将15.1g化合物6加入到反应瓶中,加入无水乙醇150ml,冰水冷却下,往反应体系中通入氯化氢气体,通入10min后,继续搅拌反应2小时。TLC分析原料反应完全,将反应液浓缩至无液体滴出。加150mL二氯甲烷到残留物中,然后再加入4.4g丁二酸酐到上述溶液中,加入11.1g三乙胺到反应液中,升温至40oC继续搅拌反应1小时,TLC分析原料反应完全,将反应液浓缩至无液体滴出,得到浅黄色粘稠液体AHU-377 13.9g,ee值为99.77%(手性HPLC分析),手性HPLC图谱见附图2。
- 还没有人留言评论。精彩留言会获得点赞!