一种调节PI3K‑AKT信号通路的非编码基因的筛选系统及筛选方法与流程
本发明属于基因工程领域,具体涉及一种调节PI3K-AKT信号通路的非编码基因的筛选系统及筛选方法。
背景技术:
磷脂酰肌醇3-激酶(PI3Ks)信号参与增殖、分化、凋亡和葡萄糖转运等多种细胞功能的调节,近年来发现,IA型PI3K和其下游分子蛋白激酶B(PKB或AKT)所组成的信号通路与人类肿瘤以及其他疾病的发生发展密切相关。该信号通路调节肿瘤细胞的增殖和存活,其活性异常不仅能导致细胞恶性转化,而且与肿瘤细胞的迁移、黏附、肿瘤血管生成以及细胞外基质的降解等相关,目前以PI3K-AKT信号通路关键分子为靶点的肿瘤治疗策略正在发展中。
AKT的失调还与心脏肥大有关:AKT直接靶向糖原合酶激酶-3(GSK-3)β,磷酸NFAT蛋白质,在调节完整心肌的肥大信号中,通过刺激NFAT核出口调节拮抗神经钙的行动。此外,AKT还参与糖尿病的发生,AKT信号的失调可导致胰岛素抗性的发展。AKT/mTOR信号通路也在神经退行性疾病中发挥作用。
IA型PI3K是由催化亚单位p110和调节亚单位p85所组成的二聚体蛋白,具有类脂激酶和蛋白激酶的双重活性。PI3K通过两种方式激活:一种是与具有磷酸化酪氨酸残基的生长因子受体或连接蛋白相互作用,引起二聚体构象改变而被激活;另一种是通过Ras和p110直接结合导致PI3K的活化。PI3K激活的结果是在质膜上产生第二信使PIP3,PIP3与细胞内含有PH结构域的信号蛋白AKT和PDK(phosphoinositidedependentkinase-1)结合,促使PDK1磷酸化AKT蛋白的Ser308导致AKT的活化。AKT还能通过PDK2(如整合素连接激酶ILK)对其Thr473的磷酸化而被激活,活化的AKT通过磷酸化作用激活或抑制其下游靶蛋白Bad、Caspase9、NF-κB、MDM2/p53、Foxo、GSK-3、FKHR、p21Cip1和p27Kip1等,进而调节细胞的增殖、分化、凋亡以及迁移等。
PI3K-AKT信号通路的活性还会被类脂磷酸酶PTEN(phosphatase and tensin homolog deleted on chromosome ten)和SHIP(SH2-containing inositol 5-phosphatase)负调节,PTEN和SHIP分别从PIP3的3′和5′去除磷酸,从而将PIP3转变成PI(4,5)P2和PI(3,4)P2而降解。
众所周知,蛋白质编码基因的转录只占全基因组转录的大约2%,其余的转录则均为非编码RNA如microRNA和lncRNA的转录,越来越多的研究表明,非编码RNA也通过多种方式参与调节PI3K-AKT信号通路,例如miR-21和miR-101可以通过直接或间接靶向PTEN,使PTEN失去活性,进而促使AKT活化;NF-κB的活化需要MALAT1参与;miR-145、Loc285194、Linc-RoR等参与了p53的基因调控,等等。
非编码RNA的数量众多,其中,到目前为止,已经有超过50,000的lncRNA被发现,这比蛋白质编码基因的数量要多得多,这也为我们人类基因组的复杂性提供了进一步证据。lncRNA是新型癌症生物标志物或治疗靶点的丰富来源,而且绝大部分lncRNA的生物学功能还是未知,对lncRNA进行功能性研究非常重要。
技术实现要素:
本发明提供了一种用于筛选调节PI3K-AKT信号通路的非编码基因的报告系统,该报告系统能简便地筛选出能够激活或抑制PI3K-AKT信号通路的非编码基因。
一种用于筛选调节PI3K-AKT信号通路的非编码基因的报告系统,该报告系统包括第一基因表达框和第二基因表达框,所述第一基因表达框包括依次相连的AKT靶标应答元件、第一启动子和Tet阻遏因子序列,所述第二基因表达框包括依次相连的Tet操纵因子序列和报告基因。
PI3K-AKT信号通路中,活化的AKT能够通过自磷酸化作用激活或抑制其下游靶蛋白(即AKT靶标),当AKT靶标被激活时,AKT靶标能够与本发明报告系统中的AKT靶标应答元件结合,从而驱使RNA聚合酶与第一启动子结合,开启Tet阻遏因子序列(即Tet阻遏因子的编码DNA)的表达,表达的Tet阻遏因子能够与Tet操纵因子序列(即Tet操纵因子的编码DNA)结合,抑制报告基因的表达;反之,当AKT被抑制或AKT靶标被抑制时,AKT靶标难以与本发明报告系统中的AKT靶标应答元件结合,Tet阻遏因子因表达量低而难以结合Tet操纵因子序列,使得报告基因高表达。如此,通过激活细胞中的非编码基因(如利用SAM文库)或抑制细胞中(如利用KO文库),并比较非编码基因被激活前后报告基因的表达水平,即可简便地了解该非编码基因对PI3K-AKT信号通路是否存在调节作用,并筛选出能激活或抑制PI3K-AKT信号通路的非编码基因。
本发明中,所述AKT靶标可以是目前为本领域技术人员所熟知的各个AKT下游靶蛋白,如Bad、Caspase9、NF-κB、MDM2/p53、Foxo、GSK-3、FKHR、p21Cip1和p27Kip1等。
本发明中,AKT靶标应答元件的拷贝数可以根据具体需要自由设置。
本发明中,第一启动子可以是完整的真核基因启动子(如RNA聚合酶II启动子),也可以是真核基因启动子的核心启动子元件,也可以是核心启动子元件中的TATA盒。
作为优选,所述报告基因为抗性基因、荧光蛋白基因中的至少一种。
作为优选,所述报告基因包括第一基因表达框和第二基因表达框,所述第一基因表达框由依次相连的AKT靶标应答元件、TATA盒、Tet阻遏因子和KRAB结构域序列(即KRAB结构域的编码DNA)组成,所述第二基因表达框由依次相连的Tet操纵因子和报告基因组成。
本发明还提供了包含所述报告系统的重组载体。
本发明还提供了一种调节PI3K-AKT信号通路的非编码基因的筛选系统,该筛选系统包括以下①和②,或者包括以下①和③:
①所述报告系统,或包含所述报告系统的重组载体;
②gRNA文库,或由所述gRNA文库与载体连接而成的gRNA载体文库;
所述gRNA文库包括针对待筛选非编码基因的多个gRNA(即singleguide RNA),每一gRNA均靶向待筛选非编码基因的启动子区域(该启动子区域一般是非编码基因第一个外显子上游~1kb的区域);
③双gRNA文库,或由所述双gRNA文库与载体连接而成的双gRNA载体文库;
所述双gRNA文库包括针对待筛选非编码基因的多个双gRNA,每一双gRNA均由依次相连的第二启动子、gRNA-a序列(即gRNA-a的编码基因)、crRNA支架序列、第三启动子和gRNA-b序列(即gRNA-b的编码基因)组成;
同一双gRNA转录的gRNA-a和gRNA-b分别与同一待筛选非编码基因上的两个靶位点互补配对,不同双gRNA转录的gRNA-a和gRNA-b中有至少一个互不相同。
作为优选,所述第二启动子和第三启动子为互不相同的聚合酶III启动子,如H1启动子或U6启动子。
gRNA文库或gRNA载体文库用于激活细胞中的非编码基因,而双gRNA文库或双gRNA载体文库则用于敲除细胞中的非编码基因。因此当采用含有gRNA文库的筛选系统时,该筛选系统宜与dCas9联用;当采用含有双gRNA文库的的筛选系统时,该筛选系统宜与hCas9联用。
其中,所述gRNA载体文库(即SAM文库)的构建方法包括以下步骤:
(1)针对待筛选非编码基因第一个外显子上游~1kb的区域,合成序列结构为“PCR扩增短序列I-gRNA-PCR扩增短序列II”的单链寡核苷酸,将针对所有待筛选非编码基因合成的单链寡核苷酸等量混合,获得混合单链寡核苷酸库;
所述PCR扩增短序列I的碱基序列为:5’-GTATGAGACCACTTGGATCC-3’;
所述PCR扩增短序列II的碱基序列为:5’-CCTTATTTTAACTTGCTATT-3’;
(2)以所述混合单链寡核苷酸库为模板,利用相应的引物进行PCR扩增,获得混合双链寡核苷酸库;
上下游引物分别与单链寡核苷酸中的PCR扩增短序列I和PCR扩增短序列II相结合;
(3)通过Gibson组装法将所述双链寡核苷酸库连入BsmB1-digested lentigRNA(MS2)-zeo载体中,获得所述gRNA载体文库。
本发明还提供了一种调节PI3K-AKT信号通路的非编码基因的筛选方法,该筛选方法采用含有上述gRNA文库的筛选系统进行,包括以下步骤:
S1:构建携带dCas9-VP64融合基因的真核细胞系;
dcas9不具备切割基因的活性,也不具备转录活性,只能结合到非编码基因的启动子区域,当dcas9与VP64融合后,VP64能够与RNA聚合酶II结合,进而激活相应非编码基因的转录。
作为优选,所述真核细胞系还携带有PMS2-p65-HSF1融合基因。该融合基因表达的PMS2-p65-HSF1融合蛋白能够进一步提高非编码基因的转录活性。
S2:将所述筛选系统中的gRNA载体文库导入所述真核细胞系中,获得稳定转导子;
S3:将包含所述报告系统的重组载体导入所述稳定转导子中,获得重组细胞系,重组细胞系经培养后,筛选出在培养前后报告基因存在差异表达的克隆;
当报告基因为抗性基因时,可以直接将重组细胞系置于含抗生素的培养液中培养,并经抗性筛选获得目标克隆。
当报告基因为荧光蛋白基因时,可以通过细胞分选的方法筛选出目标克隆。
S4:提取所述克隆的总RNA并反转录为cDNA,以cDNA为模板,利用引物扩增相应的非编码基因,通过比较各非编码基因在步骤S3培养前后的表达水平,筛选出调节PI3K-AKT信号通路的非编码基因。
利用上述筛选方法,本发明获得了五个在步骤S3培养前后存在差异表达的非编码基因。因此本发明还提供了非编码基因在激活PI3K-AKT信号通路中的应用,所述非编码基因为Hotair M1、AK023948、BC200、ESRG或IGF2-AS。
其中,AK023948能够通过提高AKT的磷酸化水平,进而激活AKT的下游通路。
本发明还提供了另一种调节PI3K-AKT信号通路的非编码基因的筛选方法,该筛选方法采用含有上述双gRNA文库的筛选系统进行,包括以下步骤:
S1’:构建携带hCas9的真核细胞系;
S2’:将所述双gRNA载体文库导入所述真核细胞系中,获得稳定转导子;
S3’:将包含所述报告系统的重组载体导入所述稳定转导子中,获得重组细胞系,重组细胞系经培养后,筛选出在培养前后报告基因存在差异表达的克隆;
S4’:提取所述克隆的总RNA并反转录为cDNA,以cDNA为模板,利用引物扩增相应的非编码基因,通过比较各非编码基因在步骤S3培养前后的表达水平,筛选出调节PI3K-AKT信号通路的非编码基因。
与现有技术相比,本发明的有益效果为:
本发明利用基于CRISPR/dCas9构建的SAM文库(即gRNA文库)激活非编码基因的转录和表达、或者利用基于CRISPR/hCas9构建的KO文库(即双gRNA文库)抑制非编码基因的转录和表达,再进一步利用报告系统相对容易地进行“大海捞针”,筛选出能够调节PI3K-AKT信号通路的有效非编码基因,从而进一步了解非编码基因的生物学功能,为调节PI3K-AKT信号通路提供新途径。
附图说明
图1为实施例1中SAM文库中gRNA与lncRNA的对应区域;
其中,Exon1表示第一个外显子;
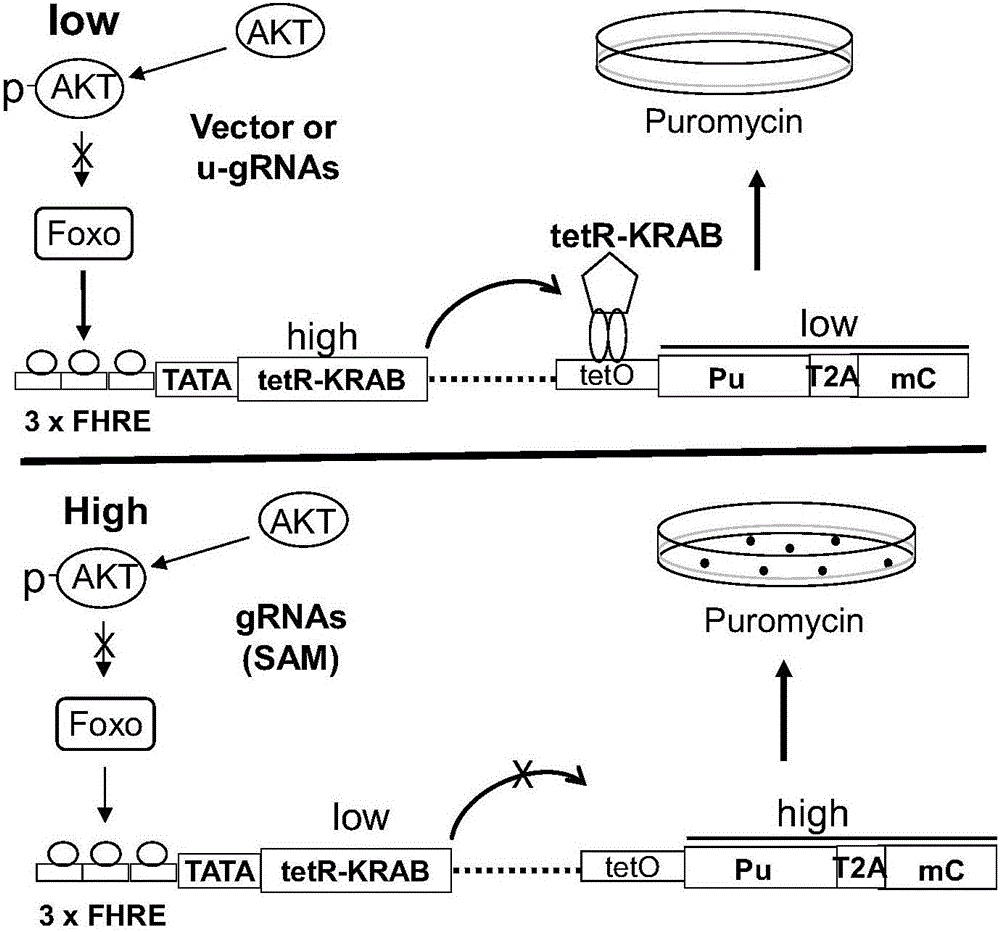
图2为本发明调节PI3K-AKT信号通路的非编码基因的筛选系统的工作原理图;
其中,high表示高,low表示低,AKT表示蛋白激酶B,p-AKT表示磷酸化的蛋白激酶B,Foxo表示Foxo转录因子,Vector or u-gRNA表示空白载体或无效的gRNA,3×FHRE表示3个拷贝数的叉头应答元件,TATA表示TATA盒,tetR-KRAB表示tet阻遏因子-KRAB融合抑制子,tetO表示tet操纵因子,Pu表示嘌呤霉素抗性基因,mC表示荧光蛋白基因mcherry,gRNA(SAM)表示有效gRNA,Puromycin表示嘌呤霉素,下同;
图3a为通过实施例2筛选获得的五个lncRNA在重组细胞系和存活克隆中的相对表达水平;
其中,Relative expression level表示相对表达水平,BS表示五个lncRNA在重组细胞系中的相对表达水平,AS表示五个lncRNA在存活克隆中的相对表达水平,下同;
图3b为针对AK023948设计的双gRNA对细胞中pAKT表达水平的影响;
图4a为AK0SAM文库和空白载体对细胞在嘌呤霉素存在下存活状况的影响;
其中,Vector表示空白载体,下同;
图4b为AK0SAM文库和空白载体对细胞中内源性AK023948表达水平的影响;
其中,Relative AK0level表示AK023948的相对表达水平,下同;
图4c为AK0SAM文库和空白载体对细胞中pAKT相对表达水平的影响;
图5a为AK023948异位表达和RNAi抑制AK023948对细胞中pAKT相对表达水平的影响;
其中,AK0表示AK023948,AK0siRNA表示采用siRNA抑制AK023948表达,Ctrl siRNA表示阴性siRNA,下同;
图5b为针对AK023948设计的双gRNA的结构示意图;
图5c为AK023948敲除对细胞中pAKT相对表达水平的影响;
其中,KO#13、KO#28、KO#32分别表示三个AK023948敲除克隆,下同;
图5d为在AK023948敲除克隆中重新表达AK023948后对细胞中pAKT相对表达水平的影响;
图6a为原位杂交染色检测AK023948和免疫组织化学染色检测pAKT的结果;
其中,ISH表示原位杂交染色,IHC表示免疫组织化学染色;
图6b为通过异种移植肿瘤的免疫组化分析AK023948敲除对细胞中pAKT相对表达水平的影响;
其中,AK0KO#13表示AK023948敲除克隆。
具体实施方式
下面结合附图和具体实施方式对本发明的技术方案作进一步详细说明。
实施例1构建调节PI3K-AKT信号通路的非编码基因的筛选系统
1SAM文库的构建
本实施例SAM文库的构建方法包括:
(1)从www.lncrnadb.org和http://www.cuilab.cn/lncrnadisease中挑选了241条来源可靠的lncRNA,并针对每条lncRNA第一个外显子上游1kb的区域(如图1)设计5个gRNA,同时针对非人类基因设计了10个gRNA作为阴性对照;
(2)与每条gRNA相对应地,委托美国customarray公司(http://www.customarrayinc.com/)合成序列结构为“PCR扩增短序列I-gRNA-PCR扩增短序列II”的单链寡核苷酸,将针对所有待筛选非编码基因合成的单链寡核苷酸等量混合,获得混合单链寡核苷酸库;
其中,PCR扩增短序列I的碱基序列为(如SEQ ID No.1698所示):
5’-GTATGAGACCACTTGGATCC-3’;
PCR扩增短序列II的碱基序列为(如SEQ ID No.1699所示):
5’-CCTTATTTTAACTTGCTATT-3’;
(3)以混合单链寡核苷酸库为模板,利用相应的引物进行PCR扩增,获得混合双链寡核苷酸库:
上下游引物分别与单链寡核苷酸中的PCR扩增短序列I和PCR扩增短序列II相结合:
上游引物:5’-GTATGAGACCACTTGGATCC-3’(如SEQ ID No.1700所示);
下游引物:5’-CCTTATTTTAACTTGCTATT-3’(如SEQ ID No.1701所示);
将PCR反应体系混匀后稍离心,分装至8个PCR管中(50μl/tube),在PCR仪中进行扩增反应,反应参数为:98℃1min,1个循环,98℃0.5min,42℃1min,72℃0.5min,循环33个,72℃4min,1个循环,4℃保存;
采用美国Zymo Research公司纯化试剂盒对PCR产物进行纯化,获得混合双链寡核苷酸库;
(4)通过Gibson组装法将双链寡核苷酸库连入BsmB1-digested lentigRNA(MS2)-zeo载体中,获得gRNA载体文库,即为SAM文库。
lncRNA与gRNA序列、qRT-PCR引物序列的对应关系如表1。
表1各lncRNA以及与之相对应的gRNA序列和qRT-PCR引物序列
2报告系统的构建
本实施例以Foxo转录因子作为AKT靶标,以FHRE(Fork Head Response Element)作为Foxo的应答元件,委托美国GeneScript公司构建报告系统,如图2所示,本实施例的报告系统以Lentivirus为载体,载体中含有两个基因表达框:FHRE-TATA-TetR-KRAB、TetO-Pu-T2A-mc。
3筛选系统
SAM文库和报告系统即组成本实施例的筛选系统。由图2可见,本实施例筛选系统的工作原理是:
当导入细胞的空白载体或无效双gRNA时,AKT活性低,pAKT水平低,Foxo能够结合到FHRE上,启动FHRE-TATA-TetR-KRAB基因表达框的转录,表达的tetR-KRAB结合到tetO上,导致Pu和mC的转录表达水平降低,细胞因难以耐受含有嘌呤霉素(Puromycin)的生长环境而死亡。而当导入细胞的是有效的双gRNA时,pAKT活性高,由pAKT引起的Foxo磷酸化将导致Foxo的亚细胞重新分配,随后快速降解,从而难以与FHRE结合、启动FHRE-TATA-TetR-KRAB基因表达框的转录,从而Pu和mC的转录表达水平升高,使得细胞因耐受含有嘌呤霉素的生长环境而得以生存下来。
实施例2调节PI3K-AKT信号通路的lncRNA的筛选方法
本实施例一种调节PI3K-AKT信号通路的lncRNA的筛选方法,包括以下步骤:
(1)构建携带dCas9-VP64融合基因和PMS2-p65-HSF1融合基因的MCF-7细胞;
具体包括以下步骤:
①在含DMEM完全培养基的六孔板中接种MCF-7细胞,接种量为5x104个/孔,培养一天;
②除去培养基,并在每孔中加入500μl含8μg聚凝胺的DMEM完全培养基、以及500μl病毒混合物(携带有MS2-dCas9-VP64融合基因和pMS2-p65-HSF1融合基因),培养一天;
③将六孔板中的培养基替换为DMEM完全培养基,继续培养至少两天;
④收集细胞,重新接种在含有新鲜DMEM完全培养基的10cm平板中,培养一天;
⑤向平板中加入10μl潮霉素(浓度为5mg/ml)和10μl杀稻瘟菌素(浓度为200mg/ml),培养一周,获得携带dCas9-VP64融合基因和PMS2-p65-HSF1融合基因的MCF-7细胞,待用。
(2)将实施例1制备的SAM文库导入MCF-7细胞中,获得稳定转导子;
具体包括以下步骤:
①在含DMEM完全培养基的六孔板中接种携带dCas9-VP64融合基因和PMS2-p65-HSF1融合基因的MCF-7细胞,接种量为5x104个/孔,培养一天;
②除去培养基,并在每孔中加入500μl含8μg聚凝胺的DMEM完全培养基、以及500μl SAM文库;
③将六孔板中的培养基替换为DMEM完全培养基,继续培养至少两天;
④收集细胞,重新接种在含有新鲜DMEM完全培养基的10cm平板中,培养一天;
⑤向平板中加入10μl博来霉素(浓度为5mg/ml),培养一周,获得携带有dCas9-VP64融合基因、PMS2-p65-HSF1融合基因和SAM文库的MCF-7细胞,待用。
(3)将已制备的报告系统导入稳定转导子中,获得重组细胞系;
具体包括以下步骤:
①在含DMEM完全培养基的六孔板中接种稳定转导子,接种量为5x104个/孔,培养一天;
②除去培养基,并在每孔中加入500μl含8μg聚凝胺的DMEM完全培养基、以及500μl报告系统,培养一天;
③将六孔板中的培养基替换为DMEM完全培养基,继续培养至少两天;
④收集细胞,重新接种在含有新鲜DMEM完全培养基的10cm平板中,培养一天,获得重组细胞系,待用。
(4)将重组细胞系分成两份,将其中一份重组细胞系置于嘌呤霉素浓度为0.5μg/ml的RPMI培养液中,于37℃下培养10天,收集存活的克隆;
(5)采用Direct-zolTM RNA MiniPrep Kit(Zymo Research,Irvine,CA)试剂盒分别提取另一份重组细胞系、以及步骤(4)中存活克隆的总RNA;
采用RevertAid Reverse Transcriptase(Fisher Scientific)和随机引物混合物(新英格兰生物实验室,伊普斯威奇,MA)将总RNA逆转录为cDNA;
以cDNA为模板,利用表1中列出的qRT-PCR引物进行扩增,比较步骤(3)重组细胞系和步骤(4)挑选得到的克隆中各lncRNA的表达水平,筛选出存在差异表达的lncRNA;
由图3a可见,本实施例从241条lncRNA中,五种potential lncRNAs对PI3K-AKT信号通路具有调节作用:Hotair M1、AK023948、BC200、ESRG和IGF2-AS。
(6)将与上述五种lncRNA相对应的gRNA重新导入携带dCas9-VP64融合基因和PMS2-p65-HSF1融合基因的MCF-7细胞中,重复步骤(2)-(5),进行复筛,复筛结果与步骤(5)的结果相同。
并且,由图3b可见,针对AK023948设计的gRNA能够提高细胞中pAKT的表达水平。
实施例3 AK023948对PI3K-AKT信号通路的调节作用研究
从筛选出的五个lncRNA中选择AK023948,进一步研究AK023948对PI3K-AKT信号通路的调节机制。
1、AK023948是PI3K-AKT信号通路的正调节因子
采用与实施例1中相应内容相同的方法,构建AK023948的SAM文库(以下简称AK0SAM文库),即含有与AK023948相对应的gRNA表达载体的质粒。
采用与实施例2中相应内容相同的方法,将AK0SAM文库和空白载体分别导入携带dCas9-VP64融合基因和PMS2-p65-HSF1融合基因的MCF-7细胞中,获得稳定转导子;将实施例1制备的、包含报告系统的重组载体导入稳定转导子中,获得重组细胞系;将重组细胞系置于嘌呤霉素浓度为0.5μg/ml的培养液中,37℃下培养10天,观察重组细胞的存活情况(如图4a),并检测重组细胞中AK023948的表达水平(如图4b)和pAKT的表达水平(如图4c)。
由图4a可见,与空白载体相比,AK0SAM文库能够提高重组细胞对嘌呤霉素的耐受性,提高重组细胞的存活率;由图4b可见,与空白载体相比,AK0SAM文库能够提高细胞内源性AK023948的表达水平;由图4c可见,与空白载体相比,AK0SAM文库能够提高重组细胞pAKT的表达水平。这表明AK023948对PI3K-AKT信号通路具有正调节作用。
2、AK023948与AKT活化的相关性
(1)RNAi抑制AK023948后细胞内pAKT水平
从Dhamarcon(Lafayette,CO)公司购买用于抑制AK023948表达的siRNA,将siRNA导入MCF-7细胞中,培养,检测细胞内pAKT的表达水平(如图5a所示)。
由图5a可见,AK023948的过表达提高了细胞内pAKT的表达水平,而通过RNAi抑制AK023948后,细胞内pAKT的表达水平则降低。
(2)双gRNA敲除AK023948后以及AK023948重新表达后细胞内pAKT水平
利用双gRNA敲除MCF-7细胞中的AK023948,具体包括以下步骤:
①利用Sap I制备线状双gRNA载体:
消化体系:双gRNA载体(参见:Nucleic Acids Res.2015Feb 18;43(3):e17.doi:10.1093/nar/gku1198.Epub 2014Nov 20)1μg,Buffer 2μL,Sap I(10units/μL)2μL,加水至100μL混合,37℃下消化3小时。
1%琼脂糖凝胶,80恒定电流30min,分离消化后的DNA(用美国Zymo Research公司DNA割胶回收试剂盒),切割线性DNA条带置于1.5mL管中,并在管中加入溶胶液(Zymo Research)(每100μg胶加300μL溶胶液),55℃下溶解约5min、或者直至胶全部溶解;转移至纯化柱,15,000rpm离心0.5min,加200μL wash buffer,15,000rpm离心0.5min;加200μL wash buffer,15,000rpm离心1min;将离心柱转移至一个新管中,加40μL水至柱中,15,000rpm离心1min,保存洗脱液(即线状双gRNA载体),用于后续克隆。
②克隆
在管中加入4μL gBlock DNA片段(委托IDT公司合成,如SEQ ID No.1702所示)、4μL线状双gRNA载体、2μL cold fusion混合酶(SBI),室温下静置5min后转移到冰上静置10min;加入25μL感受态细胞(10G购置美国Lucigen公司),在冰上放30min后42℃下水浴45s,再转移到冰上静置2~5min,加入500μL LB培养液,于37℃、250rpm下震荡培养1h,取100μL平铺在含卡纳霉素(25μg/mL)的LB平板上,在37℃孵育过夜,第二天数克隆数。
③用美国的Zymo Research公司的质粒提取试剂盒分离转化细菌中的质粒,纯化保存,获得含有针对AK023948的双gRNA的质粒。
其中,该双gRNA的结构如图5b所示,gRNA1和gRNA2的碱基序列分别如下:
AK023948-gRNA1:5’-GAGTTTTAGTCACCTATCTA-3’(如SEQ ID No.1702所示);
AK023948-gRNA2:5’-GGGTGATCCTTGTGCACGGCC-3’(如SEQ ID No.1703所示);
crRNA支架的碱基序列为:5’-GTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCT--AGTCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGCT-3’(如SEQ ID No.1704所示)。
④委托美国genescript公司构建含有hCas9和针对AK023948的双gRNA的表达载体、以及携带GFP基因和Pu基因的AK023948供载体(AK0donor vector)(每个DNA含量均为0.5μg),并利用上述重组载体对MCF-7细胞进行转染敲除。
敲除结果见图5c。由图5c可见,AK023948被敲除后,细胞内pAKT的表达水平下调。并且,由图5d可见,当在AK023948KO克隆中重新表达AK023948后,细胞内pAKT的表达水平再一次得到提升。
(3)采用乳腺癌组织微阵列(TMA)验证AK023948与pAKT之间的正相关性
以65例乳腺癌组织微阵列为样本,首先通过原位杂交(ISH)来检测TMA上的AK023948,然后用酸/醇的方法处理已做过ISH染色的TMA,以除去AK023948信号;然后再通过免疫组织化学(IHC)染色检测TMA上的pAKT。其中一例TMA检测结果如图6a,65例TMA的检测结果统计如表2。
表2 65例TMA的检测结果统计
由图6a可见,在该例TMA上,AK023948和pAKT均具有较高水平的表达。由表2可见,65例样本中有34例为AK023948和pAKT蛋白水平低表达,65例中20例AK023948和pAKT蛋白表达高。最后,异种移植肿瘤的免疫组化分析还表明,AK023948KO可引起pAKT表达显着降低(图6b),这验证了AK023948和pAKT之间存在正相关性。
- 还没有人留言评论。精彩留言会获得点赞!