一种四硼乙烷类化合物及其制备方法与流程
本发明属于化合物合成技术领域,尤其涉及一种四硼乙烷类化合物及其制备方法。
背景技术:
有机硼化合物在化学合成中具有广泛应用。现在芳基硼化合物的合成已经十分成熟,烷基硼化合物方面也有不少方法,但是对于同一个分子中含有多个硼酸酯结构的化合物的合成并不多。
目前对于多硼化合物的合成主要有以下几种方法:
1、偕多硼化合物,主要如反应式(1)~(6)所示的六种方法。
其中反应式式(1)~(2)为卡宾插入的方法:式(1)所述方法为Srebnik等人在Organometallics 2001,20,3962中报道的铂催化卡宾插入的方法,式(2)所述方法为王建波课题组在Org.Lett.,2014,16(2),pp 448–451中报道的卡宾插入的方法。
其次,式(3)为取代反应,所述方法为傅尧课题组在Org.Lett.,2014,16(24),pp 6342–6345中报道的铜催化偕二卤代物硼化的方法以及二硼甲烷衍生化的方法;
另外,反应式式(4)-(6)为不饱和键加成反应:式(4)所述方法为Shibata等人在Synlett 2009,1331中报道的铑催化炔烃双硼氢化反应,式(5)所述方法为Hall课题组在Nat.Chem.2011,3,894中报道的铜催化烯基硼的硼氢化反应,式(6)所述方法为近期由黄正课题组在J.Am.Chem.Soc.,2015,137(50),pp 15600–15603中报道的钴催化烯烃硼化的方法。
上述,反应式式(2)和式(3)均能克级以上大量合成,具有较好的实用性。
2、多硼乙烷结构化合物的合成方法:
首先,早在1964年,Walker小组就报道了1,2-二硼乙烯与四氯化二硼的加成反应,用以制备1,1,2,2-四硼乙烷,如反应式式(7)所述。随后Walter Siebert等人利用该方法合成了一些四硼乙烷衍生物,发表在Chem.Ber.1993,126,1291-1297上。然而这种方法需要严格的无水无氧操作,而且,使用的四氯二硼很不稳定,而另一个顺式乙烯基二硼化合物则是一类不易获得和储存的试剂。
之后,Walter Siebert小组首次在Eur.J.Inorg.Chem.1999,1693-1700中报道了利用铂催化炔基二硼化合物与联硼酸儿茶酚酯的加成反应实现了四硼乙烷甚至六硼乙烷的合成,如反应式式(8)所示。该方法原料1,2-二硼酸儿茶酚酯取代乙炔的合成是影响其实用性的主要原因。
最后是在2015年,由Morken小组发展了利用铂催化烯基硼与联硼酸儿茶酚酯加成合成三硼乙烷结构化合物的方法,如反应式式(9)所述,该方法发表在J.Am.Chem.Soc.2014,136,16140-16143上。但此方法只能合成三硼乙烷结构的化合物。
前人合成1,1,2,2-四硼乙烷结构化合物的方法均存在原料来源困难,使用昂贵的催化剂,合成的结构单一,合成方法实用性差等问题。因此,寻求一种操作简便,原料易得,并且能够大量合成该类化合物及其衍生物的方法是一个急需解决的问题。
技术实现要素:
有鉴于此,本发明所要解决的技术问题在于提供一种四硼乙烷类化合物及其制备方法,该制备方法具有较高的合成效率。
本发明提供了一种四硼乙烷类化合物的制备方法,包括以下步骤:
S1)将式(II)所示的二硼酸酯取代甲烷与第一强碱在低温下作用,再加入氧化剂进行反应,得到式(I)所示的四硼乙烷类化合物;
其中,为
R1、R2、R3与R4各自独立地为烷基、酯基、芳基或氢。
本发明还提供了一种四硼乙烷类化合物的制备方法,包括以下步骤:
S1)将式(II)所示的二硼酸酯取代甲烷与第一强碱在低温下作用,再加入氧化剂进行反应,得到式(I)所示的四硼乙烷类化合物;
S2)将式(I)所示的四硼乙烷类化合物与第二强碱在低温下作用,再加入含有R5结构的烷基亲电试剂进行反应,得到式(III)所示的四硼乙烷类化合物;
其中,为
R1、R2、R3与R4各自独立地为烷基、酯基、芳基或氢;
R5为烷基、酯基或芳基。
本发明还提供了一种四硼乙烷类化合物的制备方法,包括以下步骤:
S1)将式(II)所示的二硼酸酯取代甲烷与第一强碱在低温下作用,再加入氧化剂进行反应,得到式(I)所示的四硼乙烷类化合物;
S2)将式(I)所示的四硼乙烷类化合物与第二强碱在低温下作用,再加入含有R5结构的烷基亲电试剂进行反应,得到式(III)所示的四硼乙烷类化合物;
S3)将式(III)所示的四硼乙烷类化合物与第三强碱在低温下作用,再加入含有R6结构的烷基亲电试剂进行反应,得到式(IV)所示的四硼乙烷类化合物;
其中,为
R1、R2、R3与R4各自独立地为烷基、酯基、芳基或氢;
R5与R6各自独立地为烷基、酯基或芳基。
优选的,所述为以下结构中的一种:
优选的,所述第一强碱、第二强碱与第三强碱各自独立地为N,N-二异丙基氨基锂、2,2,6,6-四甲基哌啶锂、双三甲基硅基氨基锂、双三甲基硅基氨基钠与双三甲基硅基氨基钾中的一种或多种。
优选的,所述步骤S1)中的氧化剂为碘单质、溴单质、氯气、氟气、氯化碘、溴化碘、选择性氟试剂(Select-F)、N-氟代苯磺酰亚胺、三氟碘甲烷与三氟碘乙烷中的一种或多种;
所述步骤S2)中的含有R5结构的烷基亲电试剂为R5-X或R5-OH与第一磺酸类化合物形成的磺酸酯;X为卤素;
所述步骤S3)中的含有R6结构的烷基亲电试剂为R6-X′或R6-OH与第二磺酸类化合物形成的磺酸酯;X′为卤素。
优选的,所述第一磺酸类化合物与第二磺酸类化合物各自独立地为对甲苯磺酸、对溴苯磺酸、对硝基苯磺酸、苯磺酸、甲磺酸与三氟甲磺酸中的一种或多种。
优选的,所述步骤S1)、步骤S2)与步骤S3)的反应溶剂各自独立地为醚类溶剂、烃类与芳烃类溶剂中的一种或多种。
优选的,所述步骤S1)、步骤S2)与步骤S3)中的反应温度各自独立地为-10℃~30℃;反应时间各自独立地为2~24h。
本发明还提供了一种如式(I)、式(III)或式(IV)所示的四硼乙烷类化合物,
其中,为
R1、R2、R3与R4各自独立地为烷基、酯基、芳基或氢;
R5与R5各自独立地为烷基、酯基或芳基。
本发明提供了一种四硼乙烷类化合物的制备方法,包括以下步骤:S1)将式(II)所示的二硼酸酯取代甲烷与第一强碱在低温下作用,再加入氧化剂进行反应,得到式(I)所示的四硼乙烷类化合物;其中,为R1、R2、R3与R4各自独立地为烷基、酯基、芳基或氢。与现有技术相比,本发明原料易得,制备方法简单,官能团兼容性良好,能够实现大量合成,且合成效率较高。
附图说明
图1为本发明实施例1制备的1,1,2,2-四硼酸频哪醇酯乙烷的核磁共振氢谱图;
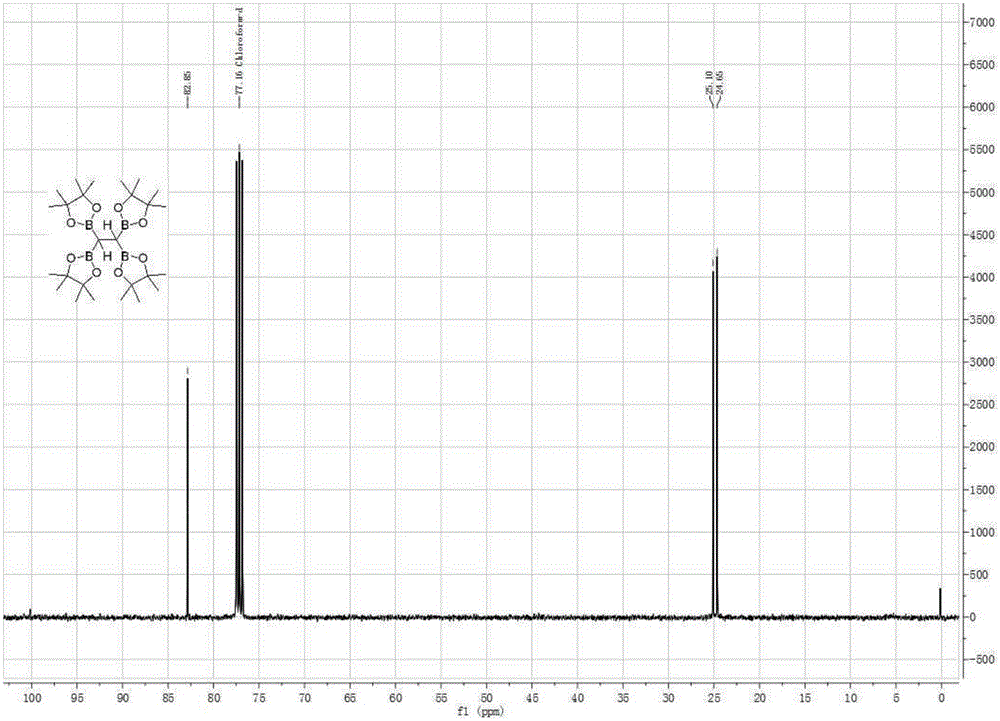
图2为本发明实施例1制备的1,1,2,2-四硼酸频哪醇酯乙烷的核磁共振碳谱图;
图3为本发明实施例1制备的1,1,2,2-四硼酸频哪醇酯乙烷的核磁共振硼谱图;
图4为本发明实施例2制备的含氰基烷基取代的1,1,2,2-四硼酸频哪醇酯乙烷的核磁共振氢谱图;
图5为本发明实施例2制备的含氰基烷基取代的1,1,2,2-四硼酸频哪醇酯乙烷的核磁共振碳谱图;
图6为本发明实施例2制备的含氰基烷基取代的1,1,2,2-四硼酸频哪醇酯乙烷的核磁共振硼谱图;
图7为本发明实施例3制备的含芳基氯和芳基醚的烷基取代的1,1,2,2-四硼酸频哪醇酯乙烷的核磁共振氢谱图;
图8为本发明实施例3制备的含芳基氯和芳基醚的烷基取代的1,1,2,2-四硼酸频哪醇酯乙烷的核磁共振碳谱图;
图9为本发明实施例3制备的含芳基氯和芳基醚的烷基取代的1,1,2,2-四硼酸频哪醇酯乙烷的核磁共振硼谱图;
图10为本发明实施例4制备的含芳基溴的烷基取代的1,1,2,2-四硼酸频哪醇酯乙烷的核磁共振氢谱图;
图11为本发明实施例4制备的含芳基溴的烷基取代的1,1,2,2-四硼酸频哪醇酯乙烷的核磁共振碳谱图;
图12为本发明实施例4制备的含芳基溴的烷基取代的1,1,2,2-四硼酸频哪醇酯乙烷的核磁共振硼谱图;
图13为本发明实施例5制备的含烷基溴取代的1,1,2,2-四硼酸频哪醇酯乙烷的核磁共振氢谱图;
图14为本发明实施例5制备的含烷基溴取代的1,1,2,2-四硼酸频哪醇酯乙烷的核磁共振碳谱图;
图15为本发明实施例5制备的含烷基溴取代的1,1,2,2-四硼酸频哪醇酯乙烷的核磁共振硼谱图;
图16为本发明实施例6制备的含稠环取代的1,1,2,2-四硼酸频哪醇酯乙烷的核磁共振氢谱图;
图17为本发明实施例6制备的含稠环取代的1,1,2,2-四硼酸频哪醇酯乙烷的核磁共振碳谱图;
图18为本发明实施例6制备的含稠环取代的1,1,2,2-四硼酸频哪醇酯乙烷的核磁共振硼谱图;
图19为本发明实施例7制备的含烷基碘取代的1,1,2,2-四硼酸频哪醇酯乙烷的核磁共振氢谱图;
图20为本发明实施例7制备的含烷基碘取代的1,1,2,2-四硼酸频哪醇酯乙烷的核磁共振碳谱图;
图21为本发明实施例7制备的含烷基碘取代的1,1,2,2-四硼酸频哪醇酯乙烷的核磁共振硼谱图。
图22为本发明实施例8制备的双烷基取代的1,1,2,2-四硼酸频哪醇酯乙烷的氢核磁共振谱图;
图23为本发明实施例8制备的双烷基取代的1,1,2,2-四硼酸频哪醇酯乙烷的碳核磁共振谱图;
图24为本发明实施例8制备的双烷基取代的1,1,2,2-四硼酸频哪醇酯乙烷的硼核磁共振谱图。
具体实施方式
本发明提供了一种四硼乙烷类化合物,如式(I)、式(III)或式(IV)所示:
其中,为
R1、R2、R3与R4各自独立地为烷基、酯基、芳基或氢,优选为C1~C30的烷基、C1~C30的酯基、C6~C30的芳基或氢,更优选为C1~C20的烷基、C1~C20的酯基、C6~C20的芳基或氢,再优选为C1~C10的烷基、C1~C10的酯基、C6~C12的芳基或氢,再优选为C1~C5的烷基、C1~C5的酯基、C6~C10的芳基或氢,最优选为C1~C3的烷基、C1~C3的酯基、C6~C8的芳基或氢。
在本发明中,优选地,所述为:
在本发明提供的一些实施例中,所述R1、R2、R3与R4均优选为甲基。
R5与R6各自独立地为烷基、酯基或芳基,优选为C1~C30的烷基、C1~C30的酯基或C6~C30的芳基,更优选为C1~C30的烷基、C1~C30的酯基或C6~C30的芳基,再优选为C1~C20的烷基、C1~C20的酯基或C6~C20的芳基,再优选为C1~C10的烷基、C1~C10的酯基或C6~C15的芳基,最优选为C1~C5的烷基、C1~C5的酯基或C6~C15的芳基。在本发明提供的一些实施例中,所述-R5与-R6各自独立地优选为以下结构中的一种:
在本发明中,如无特殊说明,所述烷基均可为直链烷基也可为支链烷基,并无特殊的限制;所述烷基可为取代的烷基也可为未取代的烷基,所述芳基可为取代的芳基也可为未取代的芳基,并无特殊的限制;所述烷基中的取代基与芳基中的取代基各自独立地优选为卤原子、氰基、卤代烷基或烷氧基。
本发明还提供了一种四硼乙烷类化合物的制备方法,包括以下步骤:
S1)将式(II)所示的二硼酸酯取代甲烷与第一强碱在低温下作用,再加入氧化剂进行反应,得到式(I)所示的四硼乙烷类化合物;
其中,为
R1、R2、R3与R4各自独立地为烷基、酯基、芳基或氢。所述R1、R2、R3与R4均同上所述,在此不再赘述。
将式(II)所示的二硼酸酯取代甲烷与第一强碱在低温下作用;其中,所述第一强碱为本领域技术人员熟知的强碱即可,并无特殊的限制,本发明中优选为N,N-二异丙基氨基锂(LDA)、2,2,6,6-四甲基哌啶锂(LTMP)、双三甲基硅基氨基锂(LHMDS)、双三甲基硅基氨基钠(NaHMDS)与双三甲基硅基氨基钾(KHMDS)中的一种或多种;所述式(II)所示的二硼酸酯取代甲烷与第一强碱的摩尔比优选为1:(0.5~3),更优选为1:(0.8~2.5),再优选为1:(1~2),最优选为1:(1~1.5);在本发明提供的一些实施例中,所述式(II)所示的二硼酸酯取代甲烷与第一强碱的摩尔比优选为1:1.2。
在本发明中,所述式(II)所示的二硼酸酯取代甲烷与第一强碱优选在低温条件下,在反应溶剂中进行作用;所述反应溶剂为本领域技术人员熟知的有机溶剂即可,并无特殊的限制,本发明中优选为醚类溶剂、烃类与芳烃类溶剂中的一种或多种,更优选为乙醚、四氢呋喃、2-甲基四氢呋喃、叔丁基甲基醚、二异丙醚、1,4-二氧六环、正己烷、苯、甲苯与二甲苯中的一种或多种;所述低温条件优选为-10℃~30℃,更优选为-10℃至室温,再优选为0℃至室温;所述作用的时间优选为2~10min,更优选为5~10min,最优选为5~6min。
再加入氧化剂进行反应;所述氧化剂为本领域技术人员熟知的氧化剂并无特殊的限制,在本发明中优选为易释放出正化合价或者分解出自由基卤素原子的化合物,更优选为碘单质、溴单质、氯气、氟气、氯化碘、溴化碘、选择性氟试剂(Select-F)、N-氟代苯磺酰亚胺、三氟碘甲烷与三氟碘乙烷中的一种或多种;所述氧化剂与式(II)所示的二硼酸酯取代甲烷的摩尔比优选为(0.5~1):1,更优选为(0.5~0.8):1,再优选为(0.5~0.7):1,最优选为0.6:1;所述氧化剂优选与反应溶剂混合后,再加入反应液中;所述反应溶剂同上所述,在此不再赘述;所述反应的温度优选为-10℃~30℃,更优选为-10℃至室温,再优选为0℃至室温;所述反应的时间优选为2~24h,更优选为2~20h,再优选为2~15h,再优选为2~10h,再优选为2~5h,最优选为2~3h。在本发明中,加入氧化剂后优选在零度以下反应5~30min后,更优选反应10~20min,再转至室温继续进行反应。
反应结束后,洗涤反应液,分出有机相,干燥、浓缩后通过柱分离,得到式(I)所示的四硼乙烷类化合物;所述洗涤反应液所用的溶液优选依次为硫代硫酸钠溶液、稀盐酸与饱和食盐水;所述柱分离流动相优选为石油醚与乙酸乙酯的混合溶剂。
在本发明中,优选地,按照以下反应式进行:
本发明提供的制备方法原料易得,方法简单,官能团兼容性良好,能够实现大量合成,且合成效率较高。
本发明还提供了另一种四硼乙烷类化合物的制备方法,包括以下步骤:
S1)将式(II)所示的二硼酸酯取代甲烷与第一强碱在低温下作用,再加入氧化剂进行反应,得到式(I)所示的四硼乙烷类化合物;
S2)将式(I)所示的四硼乙烷类化合物与第二强碱在低温下作用,再加入含有R5结构的烷基亲电试剂进行反应,得到式(III)所示的四硼乙烷类化合物;
其中,为
R1、R2、R3与R4各自独立地为烷基、酯基、芳基或氢;
R5为烷基、酯基或芳基。
所述R1、R2、R3、R4与R5及步骤S1)均同上所述,在此不再赘述。
将式(I)所示的四硼乙烷类化合物与第二强碱在低温下作用;其中,所述第二强碱为本领域技术人员熟知的强碱即可,并无特殊的限制,本发明中优选为N,N-二异丙基氨基锂(LDA)、2,2,6,6-四甲基哌啶锂(LTMP)、双三甲基硅基氨基锂(LHMDS)、双三甲基硅基氨基钠(NaHMDS)与双三甲基硅基氨基钾(KHMDS)中的一种或多种;所述式(I)所示的四硼乙烷类化合物与第二强碱的摩尔比优选为1:(0.5~3),更优选为1:(0.8~2.5),再优选为1:(1~2),最优选为1:(1~1.5);在本发明提供的一些实施例中,所述式(I)四硼乙烷类化合物与第二强碱的摩尔比优选为1:1.2。
在本发明中,所述式(I)所示的四硼乙烷类化合物与第二强碱优选在低温条件下,在反应溶剂中进行作用;所述反应溶剂为本领域技术人员熟知的有机溶剂即可,并无特殊的限制,本发明中优选为醚类溶剂、烃类与芳烃类溶剂中的一种或多种,更优选为乙醚、四氢呋喃、2-甲基四氢呋喃、叔丁基甲基醚、二异丙醚、1,4-二氧六环、正己烷、苯、甲苯与二甲苯中的一种或多种;所述低温条件优选为-10℃~30℃,更优选为-10℃至室温,再优选为0℃至室温;所述作用的时间优选为2~10min,更优选为5~10min,最优选为5~6min。
再加入含有R5结构的烷基亲电试剂进行反应;所述含有R5结构的烷基亲电试剂优选为R5-X或R5-OH与第一磺酸类化合物形成的磺酸酯;X为卤素;其中R5-X优选为一级卤代物或二级卤代物;所述R5-OH优选为一级醇或二级醇;所述第一磺酸类化合物优选为C1~C10的烷基磺酸或C6~C20的芳基磺酸,更优选为C1~C5的烷基磺酸或C6~C12的芳基磺酸,再优选为C1~C3的烷基磺酸或C6~C8的芳基磺酸;其中烷基磺酸与芳基磺酸中的烷基与芳基均可为取代的也可为未取代的;当烷基磺酸与芳基磺酸中的烷基与芳基均含有取代基时,所述取代基优选卤素或硝基;在本发明中,所述第一磺酸类化合物最优选为对甲苯磺酸、对溴苯磺酸、对硝基苯磺酸、苯磺酸、甲磺酸或三氟甲磺酸。所述含有R5结构的烷基亲电试剂与式(I)所示的四硼乙烷类化合物的摩尔比优选为(1~2):1,更优选为(1~1.8):1,再优选为(1~1.6):1,再优选为(1~1.4):1,最优选为1.2:1;所述含有R5结构的烷基亲电试剂优选与步骤S2)中的反应溶剂混合后,再加入反应液中;所述反应溶剂同上所述,在此不再赘述;所述反应的温度优选为-10℃~30℃,更优选为-10℃至室温,再优选为0℃至室温;所述反应的时间优选为2~24h,更优选为2~20h,再优选为2~15h,再优选为2~10h,再优选为2~5h,最优选为2~3h。在本发明中,加入含有R5结构的烷基亲电试剂后优选在零度以下反应5~30min后,更优选反应10~20min,再转至室温继续进行反应。
反应结束后,洗涤反应液,分出有机相,干燥、浓缩后通过柱分离,得到式(III)所示的四硼乙烷类化合物;所述洗涤反应液所用的溶液优选依次为硫代硫酸钠溶液、稀盐酸与饱和食盐水;所述柱分离流动相优选为石油醚与乙酸乙酯的混合溶剂。
在本发明中,优选地,按照以下反应式进行:
本发明提供的制备方法原料易得,方法简单,官能团兼容性良好,能够实现大量合成,且合成效率较高。
本发明还提供了另一种四硼乙烷类化合物的制备方法,包括以下步骤:
S1)将式(II)所示的二硼酸酯取代甲烷与第一强碱在低温下作用,再加入氧化剂进行反应,得到式(I)所示的四硼乙烷类化合物;
S2)将式(I)所示的四硼乙烷类化合物与第二强碱在低温下作用,再加入含有R5结构的烷基亲电试剂进行反应,得到式(III)所示的四硼乙烷类化合物;
S3)将式(III)所示的四硼乙烷类化合物与第三强碱在低温下作用,再加入含有R6结构的烷基亲电试剂进行反应,得到式(IV)所示的四硼乙烷类化合物;
其中,为
R1、R2、R3与R4各自独立地为烷基、酯基、芳基或氢;
R5与R6各自独立地为烷基、酯基或芳基。
本发明中,所述R1、R2、R3、R4、R5与R6、步骤S1)与步骤S2)均同上所述,在此不再赘述。
将式(III)所示的四硼乙烷类化合物与第三强碱在低温下作用;将式(III)所示的四硼乙烷类化合物与第三强碱在低温下作用;其中,所述第三强碱为本领域技术人员熟知的强碱即可,并无特殊的限制,本发明中优选为N,N-二异丙基氨基锂(LDA)、2,2,6,6-四甲基哌啶锂(LTMP)、双三甲基硅基氨基锂(LHMDS)、双三甲基硅基氨基钠(NaHMDS)与双三甲基硅基氨基钾(KHMDS)中的一种或多种;所述式(III)所示的四硼乙烷类化合物与第三强碱的摩尔比优选为1:(0.5~3),更优选为1:(0.8~2.5),再优选为1:(1~2),最优选为1:(1~1.5);在本发明提供的一些实施例中,所述式(III)所示的四硼乙烷类化合物与第二强碱的摩尔比优选为1:1.2。
在本发明中,所述式(I)所示的四硼乙烷类化合物与第二强碱优选在低温条件下,在反应溶剂中进行作用;所述反应溶剂为本领域技术人员熟知的有机溶剂即可,并无特殊的限制,本发明中优选为醚类溶剂、烃类与芳烃类溶剂中的一种或多种,更优选为乙醚、四氢呋喃、2-甲基四氢呋喃、叔丁基甲基醚、二异丙醚、1,4-二氧六环、正己烷、苯、甲苯与二甲苯中的一种或多种;所述低温条件优选为-10℃~30℃,更优选为-10℃至室温,再优选为0℃至室温;所述作用的时间优选为2~10min,更优选为5~10min,最优选为5~6min。
再加入含有R6结构的烷基亲电试剂进行反应;所述含有R6结构的烷基亲电试剂优选为R6-X′或R6-OH与第二磺酸类化合物形成的磺酸酯;X′为卤素,即F、Cl、Br或I;其中R6-X′优选为一级卤代物或二级卤代物;所述R6-OH优选为一级醇或二级醇;所述第二磺酸类化合物优选为C1~C10的烷基磺酸或C6~C20的芳基磺酸,更优选为C1~C5的烷基磺酸或C6~C12的芳基磺酸,再优选为C1~C3的烷基磺酸或C6~C8的芳基磺酸;其中烷基磺酸与芳基磺酸中的烷基与芳基均可为取代的也可为未取代的;当烷基磺酸与芳基磺酸中的烷基与芳基均含有取代基时,所述取代基优选卤素或硝基;在本发明中,所述第二磺酸类化合物最优选为对甲苯磺酸、对溴苯磺酸、对硝基苯磺酸、苯磺酸、甲磺酸或三氟甲磺酸。所述含有R6结构的烷基亲电试剂与式(III)所示的四硼乙烷类化合物的摩尔比优选为(1~2):1,更优选为(1~1.8):1,再优选为(1~1.6):1,再优选为(1~1.4):1,最优选为1.2:1;所述含有R5结构的烷基亲电试剂优选与步骤S3)中的反应溶剂混合后,再加入反应液中;所述反应溶剂同上所述,在此不再赘述;所述反应的温度优选为-10℃~30℃,更优选为-10℃至室温,再优选为0℃至室温;所述反应的时间优选为2~24h,更优选为2~20h,再优选为2~15h,再优选为2~10h,再优选为2~5h,最优选为2~3h。在本发明中,加入含有R6结构的烷基亲电试剂后优选在零度以下反应5~30min后,更优选反应10~20min,再转至室温继续进行反应。
反应结束后,洗涤反应液,分出有机相,干燥、浓缩后通过柱分离,得到式(IV)所示的四硼乙烷类化合物;所述洗涤反应液所用的溶液优选依次为硫代硫酸钠溶液、稀盐酸与饱和食盐水;所述柱分离流动相优选为石油醚与乙酸乙酯的混合溶剂。
在本发明中,所述步骤S1)、S2)与S3)均优选在保护气体保护的条件下进行;所述保护气体为本领域技术人员熟知的保护气体即可,并无特殊的限制,本发明中优选为氩气。
在本发明中,优选地,按照以下反应式进行:
本发明提供的制备方法原料易得,方法简单,官能团兼容性良好,能够实现大量合成,且合成效率较高。
下面将结合本发明实施例的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
实施例1 1,1,2,2-四硼酸频哪醇酯乙烷(1,1,2,2-tetrakis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)ethane):
反应式:
具体方法如下:在真空反应器内称取原料亚甲基二硼酸频哪醇酯(bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)methane)20毫摩尔,抽真空,通高纯氩气,置换三次,在氩气流保护下加入100毫升超干四氢呋喃做溶剂,适当搅拌至原料完全溶解,将容器置于零摄氏度的低温反应器中,降温至零度并保持。然后在一分钟之内,将2摩尔每升的LDA溶液12毫升,在氩气流的保护下和剧烈搅拌下加入反应体系中。待反应5~6分钟后,将预先制备好的1摩尔每升碘单质的四氢呋喃溶液12毫升,在零度和剧烈搅拌下以及氩气流保护下,滴加至反应液中。继续在零度下反应15分钟后,取出反应器至室温继续搅拌2小时。反应结束,将反应液转移至分液漏斗,加入50毫升乙醚,再依次用1摩尔每升硫代硫酸钠50毫升,1摩尔每升稀盐酸50毫升,饱和食盐水50毫升洗涤,分出有机相,干燥,浓缩,最后通过硅胶柱层析法分离(石油醚:乙酸乙酯=5:1),得无色或白色晶体即为产物。收率为87%。
利用核磁共振对实施例1中得到的产物该进行分析,得到其核磁共振氢谱图,如图1所述;得到其核磁共振碳谱图,如图2所示;得到其核磁共振硼谱图,如图3所示。由图1~图3可知,该产物的结构正确。
实施例2与烷基溴反应制备含氰基烷基取代的1,1,2,2-四硼酸频哪醇酯乙烷(4-((7,7,8,8-tetrakis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)octyl)oxy)benzonitrile):
反应式:
具体方法如下:在真空反应器内称取原料1,1,2,2-四硼酸频哪醇酯乙烷(1,1,2,2-tetrakis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)ethane)1毫摩尔,抽真空,通高纯氩气,置换三次,在氩气流保护下加入10毫升超干四氢呋喃做溶剂,适当搅拌至原料完全溶解,将容器置于零摄氏度的低温反应器中,降温至零度并保持。然后在一分钟之内,将2摩尔每升的LDA溶液0.6毫升,在氩气流的保护下和剧烈搅拌下加入反应体系中。待反应5~6分钟后,将预先制备好的1摩尔每升亲电试剂R-X(4-((6-bromohexyl)oxy)benzonitrile)的四氢呋喃溶液1.2毫升,在零度和剧烈搅拌下以及氩气流保护下,滴加至反应液中。继续在零度下反应15分钟后,取出反应器至室温继续搅拌2小时。反应结束,将反应液转移至分液漏斗,加入5毫升乙醚,再依次用1摩尔每升硫代硫酸钠5毫升,1摩尔每升稀盐酸5毫升,饱和食盐水5毫升洗涤,分出有机相,干燥,浓缩,最后通过硅胶柱层析法分离(石油醚:乙酸乙酯=5:1)得到纯的产物。收率为81%。
利用核磁共振对实施例2中得到的产物该进行分析,得到其核磁共振氢谱图,如图4所述;得到其核磁共振碳谱图,如图5所示;得到其核磁共振硼谱图,如图6所示。由图4~图6可知,该产物的结构正确。
实施例3与烷基OTs(对甲苯磺酸酯)反应制备含芳基氯和芳基醚的烷基取代的1,1,2,2-四硼酸频哪醇酯乙烷(2,2',2”,2”'-(4-(2-bromo-4-chlorophenoxy)butane-1,1,2,2-tetrayl)tetrakis(4,4,5,5-tetramethyl-1,3,2-dioxaborolane):
反应式:
具体方法如下:在真空反应器内称取原料1,1,2,2-四硼酸频哪醇酯乙烷(1,1,2,2-tetrakis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)ethane)1毫摩尔,抽真空,通高纯氩气,置换三次,在氩气流保护下加入10毫升超干四氢呋喃做溶剂,适当搅拌至原料完全溶解,将容器置于零摄氏度的低温反应器中,降温至零度并保持。然后在一分钟之内,将2摩尔每升的LDA溶液0.6毫升,在氩气流的保护下和剧烈搅拌下加入反应体系中。待反应5~6分钟后,将预先制备好的1摩尔每升亲电试剂R-X(2-(2-bromo-4-chlorophenoxy)ethyl4-methylbenzenesulfonate)的四氢呋喃溶液1.2毫升,在零度和剧烈搅拌下以及氩气流保护下,滴加至反应液中。继续在零度下反应15分钟后,取出反应器至室温继续搅拌2小时。反应结束,将反应液转移至分液漏斗,加入5毫升乙醚,再依次用1摩尔每升硫代硫酸钠5毫升,1摩尔每升稀盐酸5毫升,饱和食盐水5毫升洗涤,分出有机相,干燥,浓缩,最后通过硅胶柱层析法分离(石油醚:乙酸乙酯=10:1)得到纯的产物。收率为90%。
利用核磁共振对实施例3中得到的产物该进行分析,得到其核磁共振氢谱图,如图7所述;得到其核磁共振碳谱图,如图8所示;得到其核磁共振硼谱图,如图9所示。由图7~图9可知,该产物的结构正确。
实施例4与苄基氯反应制备含芳基溴的烷基取代的1,1,2,2-四硼酸频哪醇酯乙烷(2,2',2”,2”'-(3-(2-bromophenyl)propane-1,1,2,2-tetrayl)tetrakis(4,4,5,5-tetramethyl-1,3,2-dioxabo rolane)):
反应式:
具体方法如下:在真空反应器内称取原料1,1,2,2-四硼酸频哪醇酯乙烷(1,1,2,2-tetrakis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)ethane)1毫摩尔,抽真空,通高纯氩气,置换三次,在氩气流保护下加入10毫升超干四氢呋喃做溶剂,适当搅拌至原料完全溶解,将容器置于零摄氏度的低温反应器中,降温至零度并保持。然后在一分钟之内,将2摩尔每升的LDA溶液0.6毫升,在氩气流的保护下和剧烈搅拌下加入反应体系中。待反应5~6分钟后,将预先制备好的1摩尔每升亲电试剂R-X(1-bromo-2-(chloromethyl)benzene)的四氢呋喃溶液1.2毫升,在零度和剧烈搅拌下以及氩气流保护下,滴加至反应液中。继续在零度下反应15分钟后,取出反应器至室温继续搅拌2小时。反应结束,将反应液转移至分液漏斗,加入5毫升乙醚,再依次用1摩尔每升硫代硫酸钠5毫升,1摩尔每升稀盐酸5毫升,饱和食盐水5毫升洗涤,分出有机相,干燥,浓缩,最后通过硅胶柱层析法分离(石油醚:乙酸乙酯=10:1)得到纯的产物。收率为60%。
利用核磁共振对实施例4中得到的产物该进行分析,得到其核磁共振氢谱图,如图10所述;得到其核磁共振碳谱图,如图11所示;得到其核磁共振硼谱图,如图12所示。由图10~图12可知,该产物的结构正确。
实施例5与烷基溴反应制备含烷基溴取代的1,1,2,2-四硼酸频哪醇酯乙烷(2,2',2”,2”'-(7-bromoheptane-1,1,2,2-tetrayl)tetrakis(4,4,5,5-tetramethyl-1,3,2-dioxaborolane)):
反应式:
具体方法如下:在真空反应器内称取原料1,1,2,2-四硼酸频哪醇酯乙烷(1,1,2,2-tetrakis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)ethane)1毫摩尔,抽真空,通高纯氩气,置换三次,在氩气流保护下加入10毫升超干四氢呋喃做溶剂,适当搅拌至原料完全溶解,将容器置于零摄氏度的低温反应器中,降温至零度并保持。然后在一分钟之内,将2摩尔每升的LDA溶液0.6毫升,在氩气流的保护下和剧烈搅拌下加入反应体系中。待反应5~6分钟后,将预先制备好的1摩尔每升亲电试剂R-X(1,5-dibromopentane)的四氢呋喃溶液1.5毫升,在零度和剧烈搅拌下以及氩气流保护下,滴加至反应液中。继续在零度下反应15分钟后,取出反应器至室温继续搅拌2小时。反应结束,将反应液转移至分液漏斗,加入5毫升乙醚,再依次用1摩尔每升硫代硫酸钠5毫升,1摩尔每升稀盐酸5毫升,饱和食盐水5毫升洗涤,分出有机相,干燥,浓缩,最后通过硅胶柱层析法分离(石油醚:乙酸乙酯=10:1)得到纯的产物。收率为62%。
利用核磁共振对实施例5中得到的产物该进行分析,得到其核磁共振氢谱图,如图13所述;得到其核磁共振碳谱图,如图14所示;得到其核磁共振硼谱图,如图15所示。由图13~图15可知,该产物的结构正确。
实施例6与烷基碘反应制备含稠环取代的1,1,2,2-四硼酸频哪醇酯乙烷(2,2',2”,2”'-(4-(naphthalen-1-yl)butane-1,1,2,2-tetrayl)tetrakis(4,4,5,5-tetramethyl-1,3,2-dioxabor olane)):
反应式:
具体方法如下:在真空反应器内称取原料1,1,2,2-四硼酸频哪醇酯乙烷(1,1,2,2-tetrakis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)ethane)1毫摩尔,抽真空,通高纯氩气,置换三次,在氩气流保护下加入10毫升超干四氢呋喃做溶剂,适当搅拌至原料完全溶解,将容器置于零摄氏度的低温反应器中,降温至零度并保持。然后在一分钟之内,将2摩尔每升的LDA溶液0.6毫升,在氩气流的保护下和剧烈搅拌下加入反应体系中。待反应5~6分钟后,将预先制备好的1摩尔每升亲电试剂R-X(1-(2-iodoethyl)naphthalene)的四氢呋喃溶液1.2毫升,在零度和剧烈搅拌下以及氩气流保护下,滴加至反应液中。继续在零度下反应15分钟后,取出反应器至室温继续搅拌2小时。反应结束,将反应液转移至分液漏斗,加入5毫升乙醚,再依次用1摩尔每升硫代硫酸钠5毫升,1摩尔每升稀盐酸5毫升,饱和食盐水5毫升洗涤,分出有机相,干燥,浓缩,最后通过硅胶柱层析法分离(石油醚:乙酸乙酯=10:1)得到纯的产物。收率为82%。
利用核磁共振对实施例6中得到的产物该进行分析,得到其核磁共振氢谱图,如图16所述;得到其核磁共振碳谱图,如图17所示;得到其核磁共振硼谱图,如图18所示。由图16~图18可知,该产物的结构正确。
实施例7与烷基碘反应制备含烷基碘取代的1,1,2,2-四硼酸频哪醇酯乙烷(2,2',2”,2”'-(10-iododecane-1,1,2,2-tetrayl)tetrakis(4,4,5,5-tetramethyl-1,3,2-dioxaborolane)):
反应式:
具体方法如下:在真空反应器内称取原料1,1,2,2-四硼酸频哪醇酯乙烷(1,1,2,2-tetrakis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)ethane)1毫摩尔,抽真空,通高纯氩气,置换三次,在氩气流保护下加入10毫升超干四氢呋喃做溶剂,适当搅拌至原料完全溶解,将容器置于零摄氏度的低温反应器中,降温至零度并保持。然后在一分钟之内,将2摩尔每升的LDA溶液0.6毫升,在氩气流的保护下和剧烈搅拌下加入反应体系中。待反应5~6分钟后,将预先制备好的1摩尔每升亲电试剂R-X(1,8-diiodooctane)的四氢呋喃溶液1.5毫升,在零度和剧烈搅拌下以及氩气流保护下,滴加至反应液中。继续在零度下反应15分钟后,取出反应器至室温继续搅拌2小时。反应结束,将反应液转移至分液漏斗,加入5毫升乙醚,再依次用1摩尔每升硫代硫酸钠5毫升,1摩尔每升稀盐酸5毫升,饱和食盐水5毫升洗涤,分出有机相,干燥,浓缩,最后通过硅胶柱层析法分离(石油醚:乙酸乙酯=10:1)得到纯的产物。收率为79%。
利用核磁共振对实施例7中得到的产物该进行分析,得到其核磁共振氢谱图,如图19所述;得到其核磁共振碳谱图,如图20所示;得到其核磁共振硼谱图,如图21所示。由图19~图20可知,该产物的结构正确。
实施例8烷基OTs与实施例6的产物反应制备双烷基取代的1,1,2,2-四硼酸频哪醇酯乙烷(2,2',2”,2”'-(1-(naphthalen-1-yl)-7-(3-(trifluoromethyl)phenoxy)heptane-3,3,4,4-tetrayl)tetrakis(4,4,5,5-tetramethyl-1,3,2-dioxaborolane)):
反应式:
具体方法如下:在真空反应器内称取实施例6合成出的产物作为该反应的原料(2,2',2”,2”'-(4-(naphthalen-1-yl)butane-1,1,2,2-tetrayl)tetrakis(4,4,5,5-tetramethyl-1,3,2-dioxaborolane))0.5毫摩尔,抽真空,通高纯氩气,置换三次,在氩气流保护下加入3毫升超干四氢呋喃做溶剂,适当搅拌至原料完全溶解,将容器置于零摄氏度的低温反应器中,降温至零度并保持。然后在一分钟之内,将2摩尔每升的LDA溶液0.35毫升,在氩气流的保护下和剧烈搅拌下加入反应体系中。待反应5~6分钟后,将预先制备好的1摩尔每升亲电试剂R-X(3-(3-(trifluoromethyl)phenoxy)propyl 4-methylbenzenesulfonate)的四氢呋喃溶液0.75毫升,在零度和剧烈搅拌下以及氩气流保护下,滴加至反应液中。继续在零度下反应15分钟后,取出反应器至室温继续搅拌6小时。反应结束,将反应液转移至分液漏斗,加入5毫升乙醚,再依次用1摩尔每升硫代硫酸钠5毫升,1摩尔每升稀盐酸5毫升,饱和食盐水5毫升洗涤,分出有机相,干燥,浓缩,最后通过硅胶柱层析法分离(石油醚:乙酸乙酯=20:1)得到纯的产物。收率为38%。
利用核磁共振对实施例8中得到的产物该进行分析,得到其核磁共振氢谱图,如图22所述;得到其核磁共振碳谱图,如图23所示;得到其核磁共振硼谱图,如图24所示。由图22~图24可知,该产物的结构正确。
以上实施例的说明只是用于帮助理解本发明的方法及其核心思想。应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以对本发明进行若干改进和修饰,这些改进和修饰也落入本发明权利要求的保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!