天然双黄酮I3,II8‑Biapgienin和RidiculuflavoneA的制备方法与流程
本发明属于新化合物制备方法及药物领域,尤其是天然双黄酮化合物I3,II8-Biapgienin和RidiculuflavoneA的制备。
背景技术:
I3,II8-Biapgienin是从贯叶连翘((Hypericum perfortum L)中提取得到的一种双黄酮类化合物,贯叶连翘在欧美等国又名圣约翰草(St.John's Wort)。贯叶连翘在德国用于治疗抑郁症已有几百年历史,有报道显示I3,II8-Biapgienin具有较好的抗抑郁、抗肿瘤、抗氧化、抗炎、抗肝损伤活性,还对雌激素受体、苯二氮受体和CYP有抑制活性,在药品开发与应用方面具有广阔的前景。
Ridiculuflavone A是从一种马铃薯科的植物Aristolochia ridicula中提取的天然双黄酮化合物。由于其来源的稀少,限制了对其活性的研究。
技术实现要素:
本发明的目的在于提供两个双黄酮化合物制备方法,本发明合成出天然双黄酮化合物I3,II8-Biapgienin和Ridiculuflavone A,且合成和纯化方法简单。
本发明的目的通过以下技术方案实现的:
双黄酮化合物I3,II8-Biapgienin和Ridiculuflavone A的结构式如下:
所述衍生物为5,5',7,7'-四羟基-2,2'-双(4-羟基苯基)-4H,4'H-[3,8'-二苯并吡喃]-4,4'-二酮。
所述衍生物为2'-(3,4-二羟基苯基)-5,5',7,7'-四羟基-2-(4-羟基苯基)-4H,4'H-[3,6'-二苯并吡喃]-4,4'-二酮。
天然双黄酮I3,II8-Biapgienin的制备方法,步骤如下
天然双黄酮Ridiculuflavone A的制备方法,步骤如下:
本发明的优点和积极效果:
本发明制备的双黄酮化合物I3,II8-Biapgienin和Ridiculuflavone A具有广泛的生物活性,本发明首次实现了这两种天然产物的全合成,且合成、纯化方法简单。解决了因化合物来源稀少而对它们活性研究的限制,为其进一步开发和应用创造条件。
附图说明
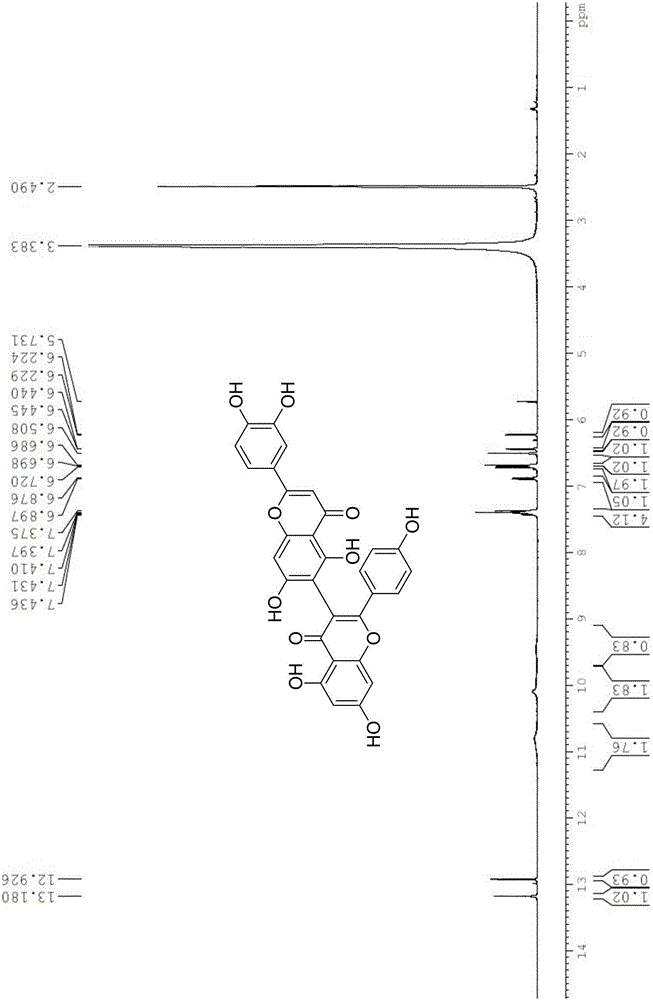
图1为5,5',7,7'-四羟基-2,2'-双(4-羟基苯基)-4H,4'H-[3,8'-二苯并吡喃]-4,4'-二酮的核磁共振氢谱。
图2为(2'-(3,4-二羟基苯基)-5,5',7,7'-四羟基-2-(4-羟基苯基)-4H,4'H-[3,6'-二苯并吡喃]-4,4'-二酮的核磁共振氢谱。
图3为(2'-(3,4-二羟基苯基)-5,5',7,7'-四羟基-2-(4-羟基苯基)-4H,4'H-[3,6'-二苯并吡喃]-4,4'-二酮的核磁共振碳谱。
具体实施方式
为了理解本发明,下面结合实施例对本发明作进一步说明;下述实施例是说明性的,不是限定性的,不能以下述实施例来限定本发明的保护范围。
本发明提供化合物1和2
本发明特别包括化合物:
(1)5,5',7,7'-四羟基-2,2'-双(4-羟基苯基)-4H,4'H-[3,8'-二苯并吡喃]-4,4'-二酮
(2)2'-(3,4-二羟基苯基)-5,5',7,7'-四羟基-2-(4-羟基苯基)-4H,4'H-[3,6'-二苯并吡喃]-4,4'-二酮
化合物1和2的合成路线
说明1
5,5',7,7'-四羟基-2,2'-双(4-羟基苯基)-4H,4'H-[3,8'-二苯并吡喃]-4,4'-二酮
取2-羟基-4,6-二甲氧基苯乙酮2.0g(0.010mol)和4-异丙氧基苯甲醛2.0g(0.012mol)加入到3mL乙醇中并搅拌5min。然后滴加6ml 50%的KOH水溶液到该反应混合物中,60℃加热,搅拌8h。冷却至室温后,将混合物倾入冰水中,用6mol/L盐酸调节pH到6,将析出的黄色沉淀抽滤,用水洗涤并从乙醇重结晶得到2'-羟基-4',6'-二甲氧基-4-异丙氧基查耳酮3.2g,为黄色结晶,产率91%。
取2'-羟基-4',6'-二甲氧基-4-异丙氧基查耳酮2.5g(7.3mmol)和碘单质0.2g(0.8mmol)加入到20mL二甲基亚砜中。将反应混合物在130℃下搅拌6小时。反应完成后,加入100ml水和50ml饱和硫代硫酸钠溶液,再用二氯甲烷(3×50mL)萃取混合物,合并有机层,并用100mL饱和食盐水洗涤,经无水硫酸钠干燥后减压浓缩。将粗产物用展开剂石油醚/乙酸乙酯=1:1,200-300目硅胶柱层析纯化纯化,得到4'-异丙氧基-5,7-二甲氧基芹菜素2g,为白色固体,产率80%。
取4'-异丙氧基-5,7-二甲氧基芹菜素1.1g(3.2mmol)和N-碘代丁二酰亚胺0.9g(4.0mmol)加入到的20mL N,N-二甲基甲酰胺中,并将所得反应液在60℃下搅拌8h。反应完成后,冷却至室温,将反应混合物倾入100mL水中,并用二氯甲烷(3×30mL)萃取,合并有机层,用饱和食盐水洗涤(3×100mL),经无水硫酸钠干燥并减压浓缩。将粗产物用展开剂石油醚/乙酸乙酯=1:1,200-300目硅胶柱层析纯化纯化,得到8-碘-4'-异丙氧基-5,7-二甲氧基芹菜素1.3g,为白色固体,产率86%。
在氩气下取8-碘-4'-异丙氧基-5,7-二甲氧基芹菜素1g(2.1mmol)、对甲氧基苯乙炔1.1g(8.58mmol)、PdCl2(PPh3)275mg(0.11mmol)和CuI 20mg(0.11mmol)加入到的4mL无水乙腈中。然后慢慢滴加1mL三乙胺。加入后,在氩气下将所得悬浮液在80℃下搅拌8h。冷却至室温后,加入20mL二氯甲烷。然后将反应悬浮液通过硅藻土过滤并用二氯甲烷洗涤,将有机相减压浓缩。将粗产物用展开剂石油醚/乙酸乙酯=1:1,200-300目硅胶柱层析纯化纯化,得到4'-异丙氧基-5,7-二甲氧基-8-((4-对甲氧基苯基)乙炔基)芹菜素0.8g,为白色固体,产率80%。
取4'-异丙氧基-5,7-二甲氧基-8-((4-对甲氧基苯基)乙炔基)芹菜素500mg(1.1mmol),2-羟基-4,6-二异丙基苯甲醛506mg(2.1mmol),[{RhCl(cod)}2]52mg(0.1mmol),1,2,3,4-四苯基-1,3-环戊二烯157mg(0.4mmol)和醋酸铜一水合物424mg(2.1mmol)加入到5mL邻二甲苯中,将所得悬浮液在氩气下加热至140℃,搅拌12h。冷却到室温后,将反应液用展开剂二氯甲烷/甲醇=100:1,200-300目硅胶柱层析纯化纯化,得到5,7-二异丙基-2'-(4-异丙氧基苯基)-5',7'-二甲氧基-2-(4-甲氧基苯基)-4H,4'H-[3,8'-二苯并吡喃]-4,4'-二酮525mg,为白色固体,产率70%。
取5,7-二异丙基-2'-(4-异丙氧基苯基)-5',7'-二甲氧基-2-(4-甲氧基苯基)-4H,4'H-[3,8'-二苯并吡喃]-4,4'-二酮100mg(0.14mmol)于25mL圆底烧瓶中,用5mL1,2-二氯乙烷溶解,置于-78℃下搅拌10min,三溴化硼709mg(2.8mmol)逐滴加入,反应液温度缓慢升高到100℃反应12h。搅拌条件下,向反应液加入过量的冰水猝灭反应,减压除去1,2-二氯乙烷,并对悬浮物抽滤,干燥得5,5',7,7'-四羟基-2,2'-双(4-羟基苯基)-4H,4'H-[3,8'-二苯并吡喃]-4,4'-二酮70mg,产率92%。
1H NMR(400MHz,Acetonitrile-d3)_δ13.05(s,1H),12.78(s,1H),7.78(s,3H),7.57(d,J=8.8Hz,2H),7.39(d,J=8.8Hz,2H),6.82(d,J=8.8Hz,2H),6.69(d,J=8.8Hz,2H),6.56(s,1H),6.54(d,J=2.0Hz,1H),6.31(d,J=2.0Hz,1H),6.28(s,1H).与文献(Schmidt B,Jaroszewski J W,Bro R,et al.Analytical Chemistry,2008,80(6):1978-87.)报道谱图数据一致。
说明2
(2'-(3,4-二羟基苯基)-5,5',7,7'-四羟基-2-(4-羟基苯基)-4H,4'H-[3,6'-二苯并吡喃]-4,4'-二酮
在氩气下取6-碘-3’,4’,5,7-四异丙基木犀草素1g(2.1mmol)、1.1g对甲氧基苯乙炔(8.58mmol)、75mg PdCl2(PPh3)2(0.11mmol)和20mg CuI(0.11mmol)加入到的4mL无水乙腈中。然后慢慢滴加1mL三乙胺。加入后,在氩气下将所得悬浮液在80℃下搅拌8h。冷却至室温后,加入20mL。然后将反应悬浮液通过硅藻土过滤并用二氯甲烷洗涤,将有机相减压浓缩。将粗产物用展开剂二氯甲烷/甲醇=100:1,200-300目硅胶柱层析纯化纯化,得到化合物6-(对甲氧苯乙炔基)-3’,4’,5,7-四异丙基木犀草素0.83g,为白色固体,产率83%。
取6-(对甲氧苯乙炔基)-3’,4’,5,7-四异丙基木犀草素450mg(0.95mmol),2-羟基-4,6-二异丙基苯甲醛454mg(1.9mmol),[{RhCl(cod)}2]47mg(0.095mmol),1,2,3,4-四苯基-1,3-环戊二烯141mg(0.38mmol)和醋酸铜一水合物570mg(2.8mmol)加入到5mL邻二甲苯中,将所得悬浮液在氩气下加热至140℃,搅拌12h。冷却到室温后,将反应液用展开剂二氯甲烷/甲醇=100:1,200-300目硅胶柱层析纯化纯化,得到2'-(3,4-二甲氧基苯基)-5,7,-二异丙氧基-5',7'-二甲氧基-2-(4-甲氧基苯基)-4H,4'H-[3,6'-二苯并吡喃]-4,4'-二酮365mg,为白色固体,产率54%。
取2'-(3,4-二甲氧基苯基)-5,7,-二异丙氧基-5',7'-二甲氧基-2-(4-甲氧基苯基)-4H,4'H-[3,6'-二苯并吡喃]-4,4'-二酮100mg(0.14mmol)于25mL圆底烧瓶中,用5mL1,2-二氯乙烷溶解,置于-78℃下搅拌10min,三溴化硼777mg(3.1mmol)逐滴加入,反应液温度缓慢升高到100℃反应12h。搅拌条件下,向反应液加入过量的冰水猝灭反应,减压除去1,2-二氯乙烷,并对悬浮物抽滤,干燥得2'-(3,4-二羟基苯基)-5,5',7,7'-四羟基-2-(4-羟基苯基)-4H,4'H-[3,6'-二苯并吡喃]-4,4'-二酮70mg,产率90%。
1H NMR(400MHz,DMSO-d6)δ13.18(s,1H),12.93(s,1H),10.81(s,2H),10.08(s,2H),9.48(s,1H),7.44–7.38(m,4H),6.89(d,J=8.4Hz,1H),6.71(d,J=8.8Hz,2H),6.69(s,1H),6.51(s,1H),6.44(d,J=2.0Hz,1H),6.23(d,J=2.0Hz,1H).13C NMR(100 MHz,Chloroform-d)δ182.11,181.06,164.78,164.44,163.44,162.79,162.01,160.23,160.00,157.80,157.21,150.25,146.22,130.15,123.56,121.93,119.55,116.52,115.66,113.84,111.25,104.63,103.86,103.58,103.36,99.32,94.11,94.00;HRMS(ESI)m/z:Calcd for C30H17O11[M-H]-553.0776;found,553.0772。
- 还没有人留言评论。精彩留言会获得点赞!