一种2,4,6‑三甲基苯乙酰氯的新合成工艺的制作方法
本发明涉及一种有机中间体的制备方法,特别涉及一种2,4,6-三甲基苯乙酰氯的新合成工艺。
背景技术:
2,4,6-三甲基苯乙酰氯是一种有机中间体,其具有广泛的用途。在光聚合材料和不饱和树脂模型中用作光敏引发剂;在塑料及油漆中用作稳定剂,可提高其耐光、耐热性能;在医药中,用作制备抗生素、抗组织胺类药物;还可用于制备涂料、染料、模塑、胶粘剂、复合纤维材料等。
在CN102633626A中公开了一种2,4,6-三甲基苯乙酰氯的合成方法:投入氯化剂(SOCl2)和N,N-二甲基甲酰胺(DMF,催化剂)至反应釜,分批次投入2,4,6三甲基苯乙酸,控制温度进行反应,反应结束,直接转入蒸馏釜升温蒸馏(高真空),采集产品2,4,6-三甲基苯乙酰氯。
在分批投入2,4,6-三甲基苯乙酸的过程,反应产生的大量的二氧化硫和氯化氢气体夹带氯化剂从投料口逃逸,损耗氯化剂,且腐蚀设备,影响现场职业卫生安全。蒸馏釜蒸馏过程,对真空度和导热油温度要求高,高温蒸馏过程,2,4,6-三甲基苯乙酰氯会焦化,若真空度变差,会加剧2,4,6-三甲基苯乙酰氯焦化,增加蒸馏残渣,减少2,4,6-三甲基苯乙酰氯收率。实际生产过程,氯化剂损耗大于3%(摩尔当量),蒸馏残渣数量偏多1.5%(质量百分比),不利于生产的节能降耗。
可以看出,现有的2,4,6-三甲基苯乙酰氯合成技术存在问题,即真空度、导热油温度和设备要求高,工艺条件苛刻,氯化剂损耗和蒸馏残渣偏多,污染环境并影响成品含量。因此,有必要研发一种2,4,6-三甲基苯乙酰氯的新合成工艺,以简化工艺条件、降低对生产设备的需求、减少蒸馏残渣、以及提高产品纯度。
技术实现要素:
为了解决上述问题,本发明人进行了锐意研究,结果发现:引入价格低廉的低沸点溶剂,改变2,4,6-三甲基苯乙酸的投料方式和次数,反应完成后蒸馏回收溶剂,即可得到纯度较高的产品。利用此工艺条件,能够使2,4,6-三甲基苯乙酰氯的纯度达到99%,无蒸馏残渣,合成工艺条件简单,对生产的设备需求低,从而完成本发明。
本发明的目的在于提供一种2,4,6-三甲基苯乙酰氯的合成工艺,该合成工艺包括以下步骤:
步骤1,在反应釜中加入溶剂Ⅰ和2,4,6-三甲基苯乙酸;
步骤2,向上述体系中加入氯化剂,然后继续反应;
步骤3,反应结束后,回收溶剂,过滤,得到2,4,6-三甲基苯乙酰氯成品。
本发明的另一目的为提供一种利用2,4,6-三甲基苯乙酰氯的合成工艺制备得到的2,4,6-三甲基苯乙酰氯,其纯度≥99%。
根据本发明提供的2,4,6-三甲基苯乙酰氯的合成工艺,具有以下有益效果:
(1)在合成工艺中引入惰性溶剂,严格控制反应物和催化剂的加入量,减少反应原料的剩余,反应完成后无需对产品进行再次纯化即可得到高纯度的产品;
(2)合成工艺反应条件温和,反应完全后在低温下回收溶剂,操作安全,且无蒸馏残渣存在,有利于得到高纯度的产品;
(3)整个合成工艺简单,使用到的溶剂价格低廉,对使用的生产设备要求不高,在易于操作的同时,降低了成本,这些因素均有利于产业化的推广;
(4)在整个合成工艺中,产生的酸性气体和反应溶剂可以直接回收利用,避免了二次污染,因此更加环保。
附图说明
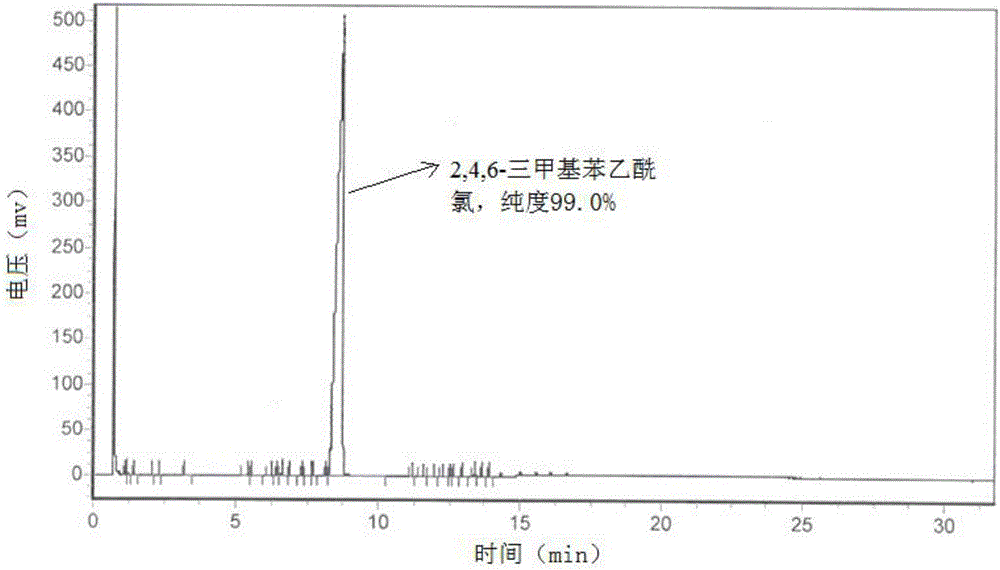
图1为实施例1中制备得到的2,4,6-三甲基苯乙酰氯的气相色谱图;
图2为实施例2中制备得到的2,4,6-三甲基苯乙酰氯的气相色谱图;
图3为对比例1中制备得到的2,4,6-三甲基苯乙酰氯的气相色谱图;
图4为对比例2中制备得到的2,4,6-三甲基苯乙酰氯的气相色谱图;
图5为对比例3中制备得到的2,4,6-三甲基苯乙酰氯的气相色谱图。
具体实施方式
下面通过对本发明进行详细说明,本发明的特点和优点将随着这些说明而变得更为清楚、明确。
根据本发明的一方面,提供一种2,4,6-三甲基苯乙酰氯的合成工艺,该合成工艺包括以下步骤:
步骤1,在反应釜中加入溶剂Ⅰ和2,4,6-三甲基苯乙酸;
步骤2,向上述体系中加入氯化剂,然后继续反应;
步骤3,反应结束后,回收溶剂,过滤,得到2,4,6-三甲基苯乙酰氯成品。
步骤1中,在反应釜中加入溶剂Ⅰ和2,4,6-三甲基苯乙酸。
在本发明中,步骤1中还加入有催化剂,催化剂可与溶剂Ⅰ一同加入或在溶剂Ⅰ后加入。
在本发明中,所述催化剂选自季铵盐类、叔胺类、季铵碱类和季膦盐类中的一种或多种,优选为叔胺类,如N,N-二甲基甲酰胺、N,N-二甲基苯胺、N,N-二乙基苯胺、4-二甲氨基吡啶和吡啶,更优选为N,N-二甲基甲酰胺。随着反应的进行,氯化氢(HCl)不断产生,催化剂N,N-二甲基甲酰胺在反应后期会在此酸性环境下发生分解,其最终不会存在或以微量残存于产品中,对产品品质影响较小。
在本发明中,溶剂Ⅰ选自氯苯、二氯苯、二氯乙烷、苯、甲苯或对二甲苯中的一种或几种以任意比例混合,优选为甲苯。上述溶剂Ⅰ在此酰氯化反应中为惰性溶剂,且沸点均低于产品。甲苯沸点为110.6℃,明显低于2,4,6-三甲基苯乙酰氯的沸点282.041℃,且高于反应温度,在反应过程中以液态存在,在反应完成后易于从反应体系中分离,回收操作简单,同时甲苯价格低廉,毒性较低,适于产业化应用,因此本发明中优选甲苯作为溶剂Ⅰ。
在本发明中,所述2,4,6-三甲基苯乙酸为一次性加入,加入过程中伴随搅拌。本发明中采用溶剂Ⅰ分散反应物,2,4,6-三甲基苯乙酸可一次性加入,不用分批次投入,简化了操作步骤。在加料过程中进行搅拌,加快了反应物的分散速度,节约了操作时间。
在本发明中,所述溶剂Ⅰ的用量为2,4,6-三甲基苯乙酸用量的20(重量)%-30(重量)%。在上述用量范围内,可满足反应要求,溶剂Ⅰ用量过多(大于30(重量)%)降低反应物浓度,可能导致反应速率下降,同时,过多的溶剂Ⅰ会造成溶剂Ⅰ回收时间的增加,在生产上增加了设备和人力成本。
在本发明中,所述催化剂的用量为2,4,6-三甲基苯乙酸用量的0.4(重量)%-0.8(重量)%。本发明合成工艺制备的产品纯度高的原因之一即为严格控制杂质的引入及反应物的剩余,若过多的加入催化剂,其可能会残存在产物中造成产物颜色不正和纯度的降低。
步骤(2)中,向上述体系中加入氯化剂,然后继续反应。
在本发明中,步骤2中,调节反应温度,向上述体系中加入氯化剂,然后升温继续反应。
所述氯化剂选自氯化亚砜(SOCl2)或双(三氯甲基)碳酸酯(C3O3Cl6)。双(三氯甲基)碳酸酯,称为三光气,又称固体光气,1个双(三氯甲基)碳酸酯分子在亲核试剂(可为本发明中催化剂N,N-二甲基甲酰胺)作用下可分解,相当于3个光气分子。三光气分子毒性低,可以准确计量,且反应条件温和,收率高,为较为理想的酰氯化试剂。
在本发明中,氯化剂为氯化亚砜时,所述氯化亚砜的摩尔量与2,4,6-三甲基苯乙酸的摩尔量之比为1.01:1-1.05:1;氯化剂为双(三氯甲基)碳酸酯时,所述双(三氯甲基)碳酸酯的摩尔量与2,4,6-三甲基苯乙酸的摩尔量之比为0.34:1-0.35:1。氯化亚砜和双(三氯甲基)碳酸酯分别与2,4,6-三甲基苯乙酸的反应如下:
由上式可知,氯化亚砜与2,4,6-三甲基苯乙酸按1:1的化学当量进行反应,双(三氯甲基)碳酸酯与2,4,6-三甲基苯乙酸按1:3的化学当量进行反应。为使反应充分进行,本发明选择两反应物中一种反应物过量,优选氯化物稍微过量。当氯化亚砜的摩尔量与2,4,6-三甲基苯乙酸的摩尔量之比大于1.05:1,或双(三氯甲基)碳酸酯的摩尔量与2,4,6-三甲基苯乙酸的摩尔量之比大于0.35:1时,体系剩余较多氯化物,造成反应物的浪费。在回收甲苯时,过量的氯化亚砜可一同排出体系,过量的双(三氯甲基)碳酸酯以气体形态形式排出体系,均不会对产品纯度产生影响。然而为保证反应完全及操作安全,氯化物不能过量太多,须严格控制氯化物与2,4,6-三甲基苯乙酸的摩尔比。
在本发明中,加入氯化剂的过程中伴随搅拌,加入氯化剂的方式为滴加的方式。随着搅拌的进行,氯化剂迅速分散在反应体系中,避免了局部浓度过大,加快反应进行。
氯化剂为双(三氯甲基)碳酸酯时将其溶于溶剂Ⅱ中再进行滴加,所述溶剂Ⅱ选自氯苯、二氯苯、二氯乙烷、苯、甲苯或对二甲苯中的一种或几种以任意比例混合,优选与使用的溶剂Ⅰ相同。
在本发明中,加入氯化剂时体系的温度为5℃-40℃,优选为10℃-35℃。伴随着氯化剂的加入,反应开始进行,在此较低温度范围内,氯化剂滴加完毕,在氯化剂滴加过程中,可完成本反应中绝大部分2,4,6-三甲基苯乙酰氯的制备。
在本发明中,加入氯化剂后升高体系的温度至50℃-65℃,优选为55℃-60℃,在较高的温度下保证反应充分、完全进行。
本发明中所述加热方式为普通加热方式,而不是回流加热方式,相较于目前广泛使用的回流加热方式,本发明对设备要求大大降低,节约了生产成本,便于大规模推广使用。
本发明人发现,升温后,保温反应的时间小于2h时,酰氯化反应进行的不够充分,体系中仍存存在未反应的原料,不仅降低了产物的收率,造成原料的浪费,同时残存的2,4,6-三甲基苯乙酸的沸点(312.9℃)高于2,4,6-三甲基苯乙酰氯(282.04℃),不易通过蒸馏分离,需要其他手段进行进一步分离提纯,造成产品纯度下降及后处理繁琐。保温反应的时间大于8h时,产物收率不再明显提高,同时浪费时间,提高了人力和设备使用成本。因此,本发明选择反应时间为2h-8h,优选为4h-6h。
本发明步骤2中,还包括对反应产生的酸性气体进行处理,所述处理方法为使酸性气体经冷凝器冷凝后依次通入水和碱液中。酸性气体经冷凝器冷凝后,使气体中夹带的氯化亚砜得到冷却分离,降低了因夹带导致的氯化亚砜的损耗,冷却得到的氯化亚砜可重新投入系统中继续反应。
经冷凝后的气体可为氯化氢和二氧化硫(氯化剂为二氯亚砜时)或氯化氢和二氧化碳(氯化剂为双(三氯甲基)碳酸酯时),氯化氢易溶于水,二氧化硫或二氧化碳在水中溶解度较小,易与碱液发生反应。因而使酸性气体先通过水中吸附掉氯化氢,再通入碱液中除去二氧化硫或二氧化碳,如此,整个反应体系中几乎无废气排放,绿色环保,吸附酸性气体的水或碱液还可用于其他用途,产生二次效益。
所述碱液选自氢氧化钠、氢氧化钾、氢氧化钙和碳酸钠中的任意一种的水溶液,优选为氢氧化钠水溶液。所述碱液价格便宜,反应能力强。其中,所述碱液的重量百分浓度为10%-20%。
步骤(3)中,采用减压蒸馏方式回收溶剂,减压蒸馏的温度为110℃-120℃,压力为500pa-600pa。
任选地,在回收溶剂前对体系进行过滤,除去微量机械杂质。
回收溶剂包括回收溶剂Ⅰ,或回收溶剂Ⅰ和溶剂Ⅱ,当使用溶剂Ⅱ且溶剂Ⅰ和溶剂Ⅱ不同时,在不同温度下分别进行回收。
在本发明中,在低压下,采用较低的温度即可完成溶剂的回收,避免了高温蒸馏过程中2,4,6-三甲基苯乙酰氯的焦化,同时工艺安全,利于生产上的节能降耗。回收后的溶剂循环套用,降低了生产成本。
溶剂回收完后,对剩余体系进行过滤,除去微量机械杂质,在不进行其它纯化操作的情况下即得到高纯度的2,4,6-三甲基苯乙酰氯产品。所述2,4,6-三甲基苯乙酰氯的纯度≥99%。
本发明合成工艺中加入低沸点溶剂,严格控制反应物和催化剂添加量,减少反应原料的剩余,在温和条件下进行反应,反应完成后在低温下回收溶剂,合成工艺安全,操作简单,无蒸馏残渣存在,可得到高纯度的产品。
本发明的另一目的为提供一种利用2,4,6-三甲基苯乙酰氯的合成工艺制备得到的2,4,6-三甲基苯乙酰氯,其纯度≥99%。
实施例
以下通过具体实例进一步描述本发明。不过这些实例仅仅是范例性的,并不对本发明的保护范围构成任何限制。
实施例1
步骤(1),在反应釜中加入166.4g甲苯和5.0g N,N二甲基甲酰胺,开启搅拌,1次性投入832g(4.7moL)2,4,6-三甲基苯乙酸,混合搅匀;
步骤(2),调节反应釜温度至15℃,滴加565.2g(4.75mol)氯化亚砜,反应产生的二氧化硫和氯化氢气体经过冷凝器后再处理,滴加完毕后,逐渐升高反应温度至55℃,保温反应5h;
步骤(3),反应结束,过滤反应体系,在真空度500Pa,温度120℃下进行甲苯的蒸馏回收,回收完毕后对剩余体系进行减压过滤,得到产品2,4,6-三甲基苯乙酰氯。
所述产品为黄色液体。对本实施例1中产品进行气相色谱检测,结果如图1所示,通过归一化法确定产品纯度为99.3%。
实施例2
步骤(1)在反应釜中加入221.2g甲苯和4.5g N,N二甲基甲酰胺,开启搅拌,1次性投入885.0g(5.0moL)2,4,6-三甲基苯乙酸,混合搅匀;
步骤(2)调节反应釜温度至15℃,滴加624.8g(5.25mol)氯化亚砜,反应产生的二氧化硫和氯化氢气体经过冷凝器后再处理,滴加完毕后,逐渐升高反应温度至60℃,保温反应4h;
步骤(3)反应结束,过滤反应体系,在真空度600Pa,温度115℃下进行甲苯的蒸馏回收,回收完毕后对剩余体系进行减压过滤,得到产品2,4,6-三甲基苯乙酰氯。
所述产品为黄色液体。对本实施例2中产品进行气相色谱检测,结果如图2所示,通过归一化法确定产品纯度为99.0%。
对比例1
步骤(1)在反应釜中加入350g甲苯和15.0g N,N二甲基甲酰胺,开启搅拌,1次性投入885.0g(5.0moL)2,4,6-三甲基苯乙酸,混合搅匀;
步骤(2)和(3)同实施例2所述。
所述产品为深黄色液体。对本对比例1中产品进行气相色谱检测,结果如图3所示,通过归一化法确定产品纯度为95.0%。
对比例2
步骤(1)和(3)中同实施例2所述。
步骤(2)调节反应釜温度至15℃,滴加624.8g(5.25mol)氯化亚砜,反应产生的二氧化硫和氯化氢气体经过冷凝器后再处理,滴加完毕后,逐渐升高反应温度至60℃,保温反应1h。
所得产品为深黄色液体。对本对比例2中产品进行气相色谱检测,结果如图4所示,通过归一化法确定产品纯度为82.2%。
对比例3
步骤(1)和(2)中同实施例2所述。
步骤(3)反应结束,过滤反应体系,在真空度600Pa,温度250℃下进行甲苯的蒸馏回收,回收完毕后对剩余体系进行减压过滤。
所得产品为深黄色液体。对本对比例3中产品进行气相色谱检测,结果如图5所示,通过归一化法确定产品纯度为94.1%。
以上结合具体实施方式和范例性实例对本发明进行了详细说明,不过这些说明并不能理解为对本发明的限制。本领域技术人员理解,在不偏离本发明精神和范围的情况下,可以对本发明技术方案及其实施方式进行多种等价替换、修饰或改进,这些均落入本发明的范围内。本发明的保护范围以所附权利要求为准。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种双[三(2‑甲基‑2‑苯基)丙基锡]衣康酸酯配合物及其制备方法与应用与流程
- 甲基硫醇锡化合物及其制备方法与流程
- 一种2,3,5-三甲基氢醌生产工艺的制作方法与工艺
- 一种双[三(2‑甲基‑2‑苯基)丙基锡]2,2′‑联苯二甲酸酯配合物及其制备方法与应用与流程
- 一种双[三(2‑甲基‑2‑苯基)丙基锡]5‑硝基间苯二甲酸酯配合物及其制备方法与应用与流程
- 一种含苯并双环酮与唑草胺的除草组合物及其应用的制作方法与工艺
- N,N,6‑三甲基‑2‑(4‑甲基苯基)咪唑并[1,2‑a]吡啶‑3‑乙酰胺的制备方法与流程
- 一种间三氟甲基苯胺中氟离子的处理方法与流程
- 一种2,4,6‑三甲基苯乙酰氯的新合成工艺的制作方法与工艺
- 制备甲基硫醇的方法与流程