一种多巯基取代芳基化合物及制备方法与流程
本发明属于高分子材料领域,具体涉及一种多磺酸取代芳基化合物的中间体及制备方法。
背景技术:
质子交换膜燃料电池(protonexchangemembranefuelcell,即pemfc)是以氢气为燃料氧气为氧化剂的高效、低污染的发电装置。pemfc主体由质子交换膜,催化剂铂(pt)、集流板和冷却板等组成。质子交换膜作为pemfc的关键部件,它起着隔离燃料和氧化剂、以及传导质子的作用,它的性能决定着燃料电池的性能。
全芳香类聚合物,因其具有良好的可用性、加工性、化学组成的多样性及在燃料电池工作环境下可预期的稳定性,而被认为是最有前途的高性能的质子交换膜材料之一。特别是磺酸取代芳基化合物如磺化聚醚醚酮(speek),磺化聚醚砜(spees)及它们的衍生材料,已成为质子交换膜材料研究的焦点。
磺酸取代芳基化合物的质子交换膜,随着磺化程度的增加,其对应的质子交换能力增强。所以,高磺化度的磺酸取代芳基化合物的质子交换膜的制备成为目前研究的热点。制备该类质子交换膜的方法主要采用相应的高磺化单体聚合的方法。目前合成所述高磺化单体的主要方法是卤代芳基化合物的磺化。如cn1654454通过发烟硫酸与1,3-二(4-氟苯甲酰基)苯在100-120℃条件下反应6-10小时磺化得到1,3-二(3-磺酸钠基-4-氟苯)甲酰基苯;cn1470505中公开以4,4’-二烷基二苯砜在75-150℃与反烟硫酸磺化得到3,3’-二磺酸-4,4’-二氯二苯砜钠盐及3,3’-二磺酸-4,4’-二氯二苯砜钠盐三种二磺化单体。
但是,采用该方法,随着单体中磺酸基的增加,所需要的磺化条件也越来越苛刻。jp200784739公开的4,4’-二氟-3,3’,5,5’-四磺酸二苯甲酮钠盐,其磺化条件则需要在密闭条件下使用50%的发烟硫酸在温度为180℃下反应24小时。此外,由于使用了50%的发烟硫酸并且在高温条件下长时间反应,整个反应过程中会产生一定的压力。由此而带来的问题就是在实际大规模生产中,磺化条件对设备的要求较高,在一定程度上增加了生产成本。此外,大量的使用过量的磺化试剂(50%发烟硫酸)也造成大量废弃固体需要处理,提高制备成本。
技术实现要素:
鉴于现有技术中的上述不足,本发明提供了一种多磺酸基取代芳基化合物的中间体,所述中间体为多巯基取代芳基化合物。该多巯基取代芳基化合物经简单氧化即可制备得到多磺酸取代芳基化合物。
本发明的目的可以通过以下方案达到:
一种多磺酸取代芳基化合物,所述多巯基取代芳基化合物,其结构如式(t)所示:
在式t中,x为酮基、砜基、直接键合、-(cf2)f-或-c(cf3)2-中的一种,f为1~5的整数中的一种;y为卤素原子或氢原子;m,n为整数,且2≤(m+n)≤8。
从化学活性的角度考虑,通式(t)中,优选如下:x为酮基、砜基或直接键合中的一种;y为f、cl或h原子;m,n为整数,且2≤(m+n)≤8。
从原料的易得程度及成本考虑,通式(t)中,更进一步优选如下:x为酮基或砜基;y为f原子;m,n为相同整数,且4≤(m+n)≤8。
本发明还提供一种上述的多磺酸取代芳基化合物的制备方法,具体方案如下:
一种上述多磺酸取代芳基化合物的制备方法,包含如下反应:
(1)溴代反应:式i-1所示的卤代芳基化合物通过溴代反应得到式i-2所示的芳基多溴代物;
(2)取代反应:对溴代反应得到的芳基多溴代物经取代反应制备得到所述的多巯基取代芳基化合物;
其中,x为酮基、砜基、直接键合、-(cf2)f-或-c(cf3)2-中的一种,f为1~5的整数中的一种;y为卤素原子或氢原子;m,n为整数,且2≤(m+n)≤8。
考虑到原料的成本,所述卤代芳基化合物,其结构如式i-1所示,其优选如下:x为酮基、砜基或直接键合中的一种;y为f、cl或h原子;m,n为整数,且2≤(m+n)≤8。
进一步考虑到反应物的活性情况,所述卤代芳基化合物,其结构如式i-1所示,更优选如下:所述式i-1中,x为酮基或砜基;y为f原子;m,n为相同整数,且4≤(m+n)≤8。
从材料后续的功能及应用领域考虑,所述卤代芳基化合物,其结构如式i-1所示,最优选如下:其中x为酮基;y为f。
本发明的多巯基取代芳基化合物的制备方法,所述的溴代反应没有特别限定。具体举例可以为,常温下,将式(i-1)所示的卤代芳基化合物溶解在硫酸和乙酸的混合溶液中,待完全溶解后,降低混合液的温度到-5~5℃之间,将溴代试剂分批加入到混合液中,然后将混合液置于冰水浴或盐水浴中,控温在0~-5℃搅拌反应0.5~2h。然后再将上述混合液,置于25~60℃之间进一步反应4~6小时,反应结束后,将反应液缓慢倒入冰水中,搅拌,过滤,水洗,干燥得白色固体,对白色固体进行重结晶便可得到式(i-2)所示的芳基多溴代物。本发明中,所述的溴代试剂可以为二溴海因、液溴或n-溴代琥铂酰亚胺中的至少一种,考虑到反应的活性及操作的难易,溴代试剂优选为二溴海因或液溴中的至少一种。本发明中,根据目标产物的不同,溴代试剂与卤代芳基化合物的摩尔比为2:1~4:1,例如目的产物为双(3,5-二溴-4-氟苯基)甲酮时,使用溴代试剂与卤代芳烃化合物的摩尔比为2:1。本发明中,上述重结晶使用的溶剂可以为丙酮、乙酸乙酯、四氢呋喃或乙腈。
考虑到两种卤素原子的选择性不同,本发明的多巯基取代芳基化合物的
制备方法中,所述的取代反应中使用的试剂优选为丁基锂和硫。
具体可以举例为:
将式(i-2)所示芳基多溴代物溶解在甲苯中,在催化剂对甲苯磺酸作用下与乙二醇或乙二醇二甲醚回流除水反应至原料消失,反应冷却后加入5%碳酸氢钠、萃取、水洗、干燥得到生成缩酮。将缩酮溶解在无水四氢呋喃中,控温在-78~90℃下缓慢加入1.6~2.7m丁基锂(正己烷溶液)并保温反应30~60分钟后,然后缓慢升温至20~40℃反应2~5h。再次降温至-78~-90℃加入试剂硫粉,缓慢升温到0℃搅拌反应30-60分钟。停止反应并自然升至室温,加入1n盐酸淬灭,甲苯萃取、水洗、无水硫酸镁干燥、脱溶、纯化得到式(t)所示的多巯基取代芳基化合物。
本发明还提供一种由上述多巯基取代芳基化合物制备多磺酸取代芳基化合物的方法:
本发明的多磺酸取代芳基化合物的制备方法,包含通过上述式t所示多巯基取代芳基化合物通过氧化反应得到式i-3所示多磺酸取代芳基化合物的步骤:
其中,x为酮基、砜基、直接键合、-(cf2)f-或-c(cf3)2-中的一种,f为1~5的整数中的一种;y为卤素原子或氢原子;m,n为整数,且2≤(m+n)≤8。
考虑到原料的成本及后续材料的性能,x优选为酮基、砜基或直接键合中的一种;y为f、cl或h原子;m,n为整数,且2≤(m+n)≤8。
所述氧化反应没有特别限定,具体举例可以为,将式可以为式(t)所示的多巯基取代芳基化合物溶解在甲醇和氯仿的混合液中加热,加入10~40倍当量的氧化剂,常温下搅拌反应20h~40h。待反应结束后,脱除溶剂得到磺酸化合物粗品,溶入水中加入naoh中和,再经重结晶纯化得到相应的多磺酸钠芳基化合物。所述重结晶试剂主要可以为丙酮、四氢呋喃、氯仿、甲醇、乙酸乙酯或异丙醇等,考虑到成本价及效果,所述重结晶试剂优选丙酮或异丙醇。所述氧化反应中使用的氧化试剂为双氧水、或双氧水和三氟乙酸的混合物。
利用该多磺酸取代芳基化合物制备磺酸取代芳基化合物,整个反应过程中不需要使用强氧化试剂,反应条件温和,对设备要求不高,操作简便。此外,反应结束也无需处理大量的废酸,降低了制备成本。利用该多磺酸取代芳基化合物可聚合得到高磺化度的预聚物,可进一步制备得到优良的性能的质子膜燃料电池电解质膜。广泛应用于新型能源电池材料研究开发领域。
附图说明
图1:实施例1中双(3,5-二溴-4-氟苯基)甲酮的核磁谱图;
图2:实施例1中双(3,5-二溴-4-氟苯基)甲酮的质谱图;
图3:实施例1中双(4-氟-3,5-二巯基苯)甲酮核磁谱图;
图4:实施例10中4,4’-二氟-3,3’,5,5’-四磺酸钠二苯甲酮核磁谱图。
具体实施方式
本发明可通过下面优选实施方案的实施例获得,但这些实施例仅在于举例说明,不对本发明的范围做出界定。
实施例中使用的原材料:
实施例中使用的有机溶剂四氢呋喃、丙酮、乙醇、甲醇、氯仿、甲苯均为分析纯(ar级),购自国药集团化学试剂有限公司;去离子水是通过离子交换树脂树脂自制。分析测试中使用试剂乙腈(hplc级)、甲醇(lc-ms级)及水(lc-ms级)。原料4,4’-二氟-二苯甲酮购自常州金坛春风化工有限公司。
实施例中所用到的检测方法:
1.样品结构表征:
1)核磁(1hnmr)
采用jeol400mhz核磁进行测定,测定用的溶剂选用氘代二甲亚砜、氘代氯仿、氘代甲醇或氘代丙酮。取样品15~20mg常温溶解于0.5ml的氘代试剂中,置于核磁管中,进样扫描8~20次即可。
2)液质(lc-ms)分析
美国waters公司的超高效液相色谱-四极杆飞行时间质谱联用仪(仪器型号:acquitytmuplc&q-tofmspremier)。
取样品1~5mg,使用甲醇(lc-ms级)震荡溶解,经0.5um有机微膜过滤至2.5ml进样瓶中。使用以下测试方法,截取主峰进入ms分析,经软件分析得到对应物质的分子量及裂分数据,进而判定目标化合物的归属。(色谱柱:shimpackvp-ods4.6mm*150mm*5um;柱温:45℃;流动相:a=水、b=甲醇;流速:0.5ml/分钟;洗脱:100%-甲醇;检测器:紫外可见254nm吸收)。
2.纯度测定
(1)式(i-2)所示芳基多溴代物样品的含量测定:仪器:岛津lc-20a;色谱柱:shimpackvp-ods4.6mm*150mm*5um;柱温:30℃;流动相:a=水、b=甲醇;流速:0.5ml/分钟;洗脱:30%-水+70%-甲醇;检测器:uv254(紫外可见254nm吸收)。
(2)多磺酸取代芳基化合物的含量测定:仪器:岛津lc20a;色谱柱:tsk-gelamide-804.6×250nm,5um;柱温:30℃;流动相:a=水、b=乙腈;流速:1ml/分钟;梯度洗脱程序:0→5分钟b=98%;5→20分钟b:98→60%;20→25分钟b:60%;25→36分钟b:98%。检测器:uv254(紫外可见254nm吸收)。
(3)收率计算:收率=(产物hplc测得含量)*(固体物质的质量)/(理论上全部转化时得到的产物质量)。
实施例1
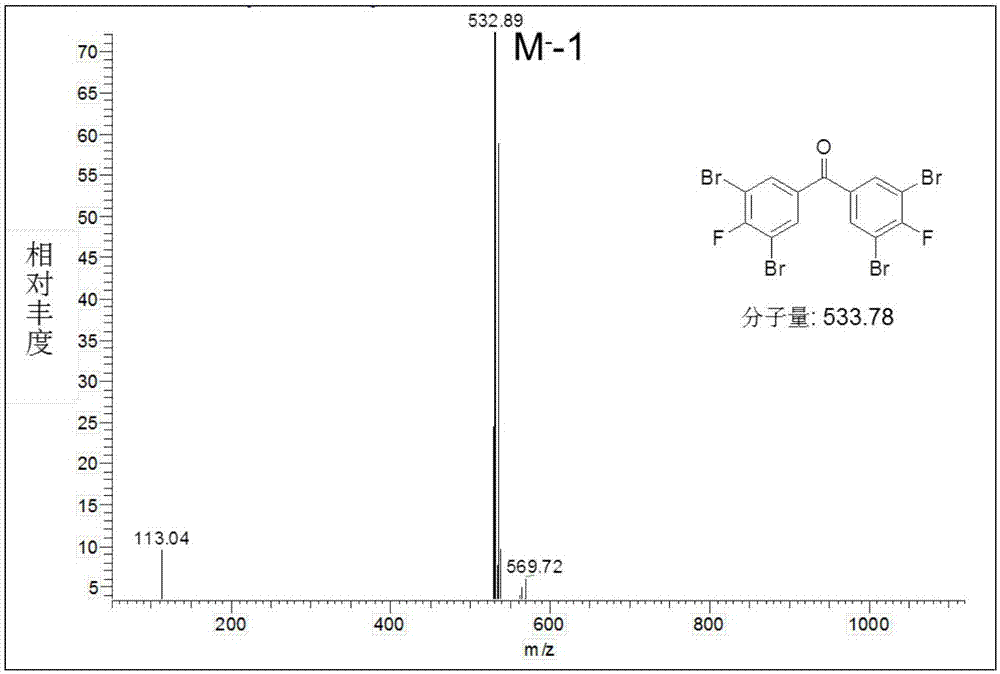
双(4-氟-3,5-二巯基苯)甲酮的合成:(1)溴代反应:将10.5g(0.048mol)的4,4’-二氟-二苯甲酮溶解在20ml乙酸和100ml浓硫酸中,常温下搅拌,20min后溶解完全;降低混合液的温度到0~-5℃之间。称取二溴海因36g(0.126mol)分批加入到上述混合液中,加入完全后,保温0℃左右搅拌2h,缓慢升温至45℃反应4h,hplc检测产物生成情况。反应结束后,将反应液缓慢倒入1kg冰水中,搅拌,过滤,水洗3*300ml,得白色固体双(3,5-二溴-4-氟苯基)甲酮。于真空干燥箱干燥2h后,使用四氢呋喃溶解结晶得到无色针状晶体19.5g,hplc测得纯度为93%,收率75%,1hnmr结构表征结果:400mhz,cdcl3,ar-h:(δ=7.915,d,j=5.48,4h);lc-ms分析得m-=532.89(理论分子量=533.78gmol-1)。(2)取代反应:双(3,5-二溴-4-氟苯基)甲酮5.3g(0.01mol)溶解在0.2l甲苯中,加入催化剂一水合对甲苯磺酸0.1g(0.5mol),乙二醇0.68g(0.011mol)于装有分水器的回流装置中加热回流6天,检测反应完全。反应冷却后加入5%碳酸氢钠200ml、萃取、水洗(3*200ml)、真空干燥得到双(3,5-二溴-4-氟苯基)缩酮5.1g,收率88%。将上述缩酮2.9g(0.005mol)溶解在100ml无水四氢呋喃中,控温在-78℃下缓慢加入2.7m丁基锂(正己烷溶液)8.0ml并保温反应30分钟后,然后缓慢升温至20℃反应2h。再次降温至-78℃加入硫粉0.7g,缓慢升温到0℃搅拌反应30min。停止反应并自然升至室温,加入1n盐酸淬灭,甲苯萃取、水洗、无水硫酸镁干燥、脱溶,得到化合物双(4-氟-3,5-二巯基苯)甲酮1.2g,收率69%。
如图1所示:给出了双(3,5-二溴-4-氟苯基)甲酮中的氢的归属及图2质谱的分子量结果,1hnmr结构表征结果:400mhz,dmso-d6,δ=7.915,d,j=5.48;lc-ms分析得m-=532.89(理论分子量=533.78gmol-1),证实了上述制备的中间体双(3,5-二溴-4-氟苯基)甲酮的结构。
如图3所示:给出了目标化合物中的氢的归属。1hnmr结构表征结果:400mhz,dmso-d6,δ1-h=7.9014,4h,δ2-h=4.9002,4h;证实了上述制备的多磺酸化合物中间体双(4-氟-3,5-二巯基苯)甲酮的结构。
实施例2
双(4-氟-3,5-二巯基苯)甲酮的合成:(1)溴代反应:将10.5g(0.048mol)的4,4’-二氟-二苯甲酮溶解在20ml乙酸和100ml浓硫酸中,常温下搅拌,20min后溶解完全;降低混合液的温度到5~0℃之间。取液溴22.7g(0.126mol)分批滴加入到上述混合液中,加入完全后,保温0℃左右搅拌2h,缓慢升温至45℃反应4h,hplc检测产物生成情况。反应结束后,将反应液缓慢倒入1kg冰水中,搅拌,过滤,水洗3*300ml,得白色固体双(3,5-二溴-4-氟苯基)甲酮。于真空干燥箱干燥2h后,使用四氢呋喃溶解结晶得到无色针状晶体19.5g,hplc测得纯度为93%。(2)取代反应:双(3,5-二溴-4-氟苯基)甲酮5.3g(0.01mol)溶解在0.2l甲苯中,加入催化剂一水合对甲苯磺酸0.1g(0.5mol),乙二醇0.68g(0.011mol)于装有分水器的回流装置中加热回流6天,检测反应完全。反应冷却后加入5%碳酸氢钠200ml、萃取、水洗(3*200ml)、真空干燥得到双(3,5-二溴-4-氟苯基)缩酮5.2g,收率91%。将上述缩酮2.9g(0.005mol)溶解在100ml无水四氢呋喃中,控温在-78℃下缓慢加入2.7m丁基锂(正己烷溶液)8.0ml并保温反应30分钟后,然后缓慢升温至20℃反应2h。再次降温至-78℃加入硫粉0.7g,缓慢升温到0℃搅拌反应30min。停止反应并自然升至室温,加入1n盐酸淬灭,甲苯萃取、水洗、无水硫酸镁干燥、脱溶,得到化合物双(4-氟-3,5-二巯基苯)甲酮1.5g,收率73%。
实施例3
双(4-氟-3,5-二巯基苯)甲酮的合成:(1)溴代反应:将10.5g(0.048mol)的4,4’-二氟-二苯甲酮溶解在200ml四氢呋喃中,常温下搅拌溶解完全;降低混合液的温度到0~-3℃之间。称取n溴代-琥珀酸亚胺44.8g(0.25mol)分批加入到上述混合液中,加入完全后,保温0℃左右搅拌2h,缓慢升温至45℃反应4h,hplc检测产物生成情况。反应结束后,将反应液缓慢倒入1kg冰水中,搅拌,过滤,水洗3*300ml,得白色固体双(3,5-二溴-4-氟苯基)甲酮。于真空干燥箱干燥2h后,使用四氢呋喃溶解结晶得到无色针状晶体16.1g。(2)取代反应:双(3,5-二溴-4-氟苯基)甲酮5.3g(0.01mol)溶解在0.2l甲苯中,加入催化剂一水合对甲苯磺酸0.1g(0.5mol),乙二醇0.68g(0.011mol)于装有分水器的回流装置中加热回流6天,检测反应完全。反应冷却后加入5%碳酸氢钠200ml、萃取、水洗(3*200ml)、真空干燥得到双(3,5-二溴-4-氟苯基)缩酮4.8g,收率81%。将上述缩酮2.9g(0.005mol)溶解在100ml无水四氢呋喃中,控温在-78℃下缓慢加入2.7m丁基锂(正己烷溶液)8.0ml并保温反应30分钟后,然后缓慢升温至20℃反应2h。再次降温至-78℃加入硫粉0.7g,缓慢升温到0℃搅拌反应30min。停止反应并自然升至室温,加入1n盐酸淬灭,甲苯萃取、水洗、无水硫酸镁干燥、脱溶,得到化合物双(4-氟-3,5-二巯基苯)甲酮1.1g,收率66%。
实施例4
双(4-氟-2,3,5-三巯基苯)甲酮的合成:(1)溴代反应:将10.5g(0.048mol)的4,4’-二氟-二苯甲酮溶解在200ml四氢呋喃中,常温下搅拌溶解完全;降低混合液的温度到2~0℃之间。称取二溴海因41.0g(0.144mol)分批加入到上述混合液中,加入完全后,保温0℃左右搅拌2h,缓慢升温至45℃反应4h,hplc检测产物生成情况。反应结束后,将反应液缓慢倒入1.3kg冰水中,搅拌,过滤,水洗3*350ml,得白色固体双(2,3,5-三溴-4-氟苯基)甲酮。于真空干燥箱干燥2h后,使用四氢呋喃溶解结晶得到无色针状晶体19.2g。(2)取代反应:双(2,3,5-三溴-4-氟苯基)甲酮5.9g(10mmol)溶解在0.2l甲苯中,加入催化剂一水合对甲苯磺酸0.1g(0.5mol),乙二醇0.68g(11mmol)于装有分水器的回流装置中加热回流6天,检测反应完全。反应冷却后加入5%碳酸氢钠200ml、萃取、水洗(3*200ml)、真空干燥得到双(2,3,5-三溴-4-氟苯基)缩酮5.4g,收率84%。将上述缩酮2.0g(2.7mmol)溶解在100ml无水四氢呋喃中,控温在-78℃下缓慢加入2.7m丁基锂(正己烷溶液)10.0ml并保温反应30分钟后,然后缓慢升温至20℃反应2h。再次降温至-78℃加入硫粉1.0g,缓慢升温到0℃搅拌反应30min。停止反应并自然升至室温,加入1n盐酸淬灭,甲苯萃取、水洗、无水硫酸镁干燥、脱溶,得白色固体粉末,进一步纯化得化合物双(4-氟-2,3,5-三巯基苯)甲酮0.6g,收率55%。
实施例5
双(4-氟-2,3,5,6-四巯基苯)甲酮的合成:(1)溴代反应:将5.25g(0.024mol)的4,4’-二氟-二苯甲酮溶解在10ml乙酸和80ml浓硫酸中,常温下搅拌溶解完全;降低混合液的温度到0~-2℃之间。称取二溴海因28.5g(0.1mol)分批加入到上述混合液中,加入完全后,保温0℃左右搅拌2h,缓慢升温至45℃反应10h,hplc检测产物生成情况。反应结束后,将反应液缓慢倒入0.8kg冰水中,搅拌,过滤,水洗3*350ml,得白色固体双(2,3,5,6-四溴-4-氟苯基)甲酮。于真空干燥箱干燥2h后,使用四氢呋喃溶解结晶得到无色针状晶体17.2g,hplc测得纯度为95%,收率84%。(2)取代反应:将双(2,3,5,6-四溴-4-氟苯基)甲酮8.5g(0.01mol)溶解在200ml甲苯中,加入催化剂一水合对甲苯磺酸0.1g(0.5mol),乙二醇0.68g(11mmol)于装有分水器的回流装置中加热回流6天,检测反应完全。反应冷却后加入5%碳酸氢钠200ml、萃取、水洗(3*200ml)、真空干燥得到双(2,3,5,6–四溴-4-氟苯基)缩酮17.4g,收率86%。将上述缩酮2.0g(2.2mmol)溶解在100ml无水四氢呋喃中,控温在-78℃下缓慢加入2.7m丁基锂(正己烷溶液)14.0ml并保温反应30分钟后,然后缓慢升温至20℃反应2h。再次降温至-78℃加入硫粉1.5g,缓慢升温到0℃搅拌反应30min。停止反应并自然升至室温,加入1n盐酸淬灭,甲苯萃取、水洗、无水硫酸镁干燥、脱溶,得白色固体粉末,进一步纯化得化合物双(4-氟-2,3,5,6-四巯基苯)甲酮0.48g,收率50%。
实施例6
4,4’-二氟-3,3’,5,5’-四磺酸钠二苯甲酮的合成:取上述实施例1制备的双(4-氟-3,5-二巯基苯)甲酮1.15g(3.3mmol)加入氯仿10ml甲醇10ml搅拌至溶解完全,加入氧化剂30%h2o2(27.2ml,0.264mol)控温(25℃)反应36h。脱除溶剂得到磺酸化合物粗品,溶入水中加入naoh中和,再经异丙醇重结晶纯化得到相应的多磺酸芳基化合物4,4’-二氟-3,3’,5,5’-四磺酸钠二苯甲酮1.48g,收率83%,1hnmr结构表征结果:400mhz,dmso-d6,ar-h:(δ=8.05,d,j=6.88,4h)如图4所示:给出了磺化产物结构中氢的归属,证实得到了对应的四磺化取代的产物。
实施例7
4,4’-二氟-3,3’,5,5’-四磺酸钠二苯甲酮的合成:取上述实施例2制备的双(4-氟-3,5-二巯基苯)甲酮1.15g(3.3mmol)加入氯仿10ml甲醇10ml搅拌至溶解完全,加入氧化剂30%h2o2(13.6ml,0.132mol)及三氟乙酸0.5ml(6.6mmol),控温(25℃)反应36h。脱除溶剂得到磺酸化合物粗品,溶入水中加入naoh中和,再经丙酮重结晶纯化得到相应的多磺酸芳基化合物4,4’-二氟-3,3’,5,5’-四磺酸钠二苯甲酮1.78g,收率86%。
实施例8
4,4’-二氟-3,3’,5,5’-四磺酸钠二苯甲酮的合成:取上述实施例3制备的双(4-氟-3,5-二巯基苯)甲酮1.15g(3.3mmol)加入氯仿10ml甲醇10ml搅拌至溶解完全,加入氧化剂30%h2o2(27.2ml,0.264mol)控温(25℃)反应36h。脱除溶剂得到磺酸化合物粗品,溶入水中加入naoh中和,再经异丙醇重结晶纯化得到相应的多磺酸芳基化合物4,4’-二氟-3,3’,5,5’-四磺酸二苯甲酮1.56g,收率84%。
实施例9
4,4’-二氟-2,2’,3,3’,5,5’-六磺酸钠二苯甲酮的合成:取上述实施例4制备的双(4-氟-2,3,5-三巯基苯)甲酮1.35g(3.3mmol)加入氯仿15ml甲醇15ml搅拌至溶解完全,加入氧化剂30%h2o2(54.4ml,0.528mol)控温(25℃)反应32h。脱除溶剂得到磺酸化合物粗品,溶入水中加入naoh中和,再经异丙醇重结晶纯化得到相应的多磺酸芳基化合物4,4’-二氟-2,2’,3,3’,5,5’-六磺酸钠二苯甲酮2.03g,收率88%。
实施例10
4,4’-二氟-2,2’,3,3’,5,5’,6,6’-八磺酸钠二苯甲酮的合成:取上述实施例5制备的双(4-氟-2,3,5,6-四巯基苯)甲酮1.56g(3.3mmol)加入氯仿25ml甲醇25ml搅拌至溶解完全,加入氧化剂30%h2o2(54.4ml,0.528mol)控温(25℃)反应32h。脱除溶剂得到磺酸化合物粗品,溶入水中加入naoh中和,再经异丙醇重结晶纯化得到相应的多磺酸芳基化合物4,4’-二氟-2,2’,3,3’,5,5’,6,6‘-八磺酸钠二苯甲酮1.87g,收率70%。
实施例11
4,4’-二氯-3,3’,5,5’-四磺酸钠二苯甲酮合成:取上述实施例1制备的双(4-氯-3,5-二巯基苯)甲酮1.25g(3.3mmol)加入氯仿10ml甲醇10ml搅拌至溶解完全,加入氧化剂30%h2o2(27.2ml,0.264mol)控温(25℃)反应36h。脱除溶剂得到磺酸化合物粗品,溶入水中加入naoh中和,再经异丙醇重结晶纯化得到相应的多磺酸芳基化合物4,4’-二氯-3,3’,5,5’-四磺酸钠二苯甲酮1.8g,收率94%。
实施例12
4,4’-二氟-3,3’,5-三磺酸钠二苯甲酮的合成:取上述实施例9制备的双(4,4’-二氟-3,3’,5-三巯基苯)甲酮1.04g(3.3mmol)加入氯仿15ml甲醇15ml搅拌至溶解完全,加入氧化剂30%h2o2(54.4ml,0.528mol)三氟乙酸(60.2g,0.528mol)控温(25℃)反应32h。脱除溶剂得到磺酸化合物粗品,溶入水中加入naoh中和,再经异丙醇重结晶纯化得到相应的多磺酸芳基化合物4,4’-二氟-3,3’,5-三磺酸钠二苯甲酮1.16g,收率77%。
对比例1
根据日本特开第2007-84739号所述的方法,合成4,4’-二氟-3,3’,5,5’-四磺酸钠二苯甲酮。即将109.1克4,4‘-二氟二苯基酮、150毫升发烟硫酸(50%so3)、50克三氧化硫在180℃反应24小时。然后将反应物逐渐地置入大量水中,将其以naoh中和,然后加入200克氯化钠而沉淀合成产物。将生成沉淀滤除而实行纯化。4,4‘-二氟而苯基酮的二磺化产物为主要产物,且确认有少量三磺化及四磺化产物。如日本特开第2007-84739号所述,仅获得二磺化产物、三磺化产物及四磺化产物的混合物,无法严格地选定相对双酚之摩尔量,且使用此单体混合物则难以获得高分子量产物。
对比例2
根据中国专利cn103814062a所述的方法,100g4,4’-二氟二苯基酮,210毫升发烟硫酸(60wt%),继而将氮气强力通入连接浓缩器上部的氮导管和导向体系外部的鼓泡器,在200℃反应24小时。此时剧烈流动的氮气抑制三氧化硫挥发。将反应溶液逐渐地置入大量水中且以naoh中和之后,用乙醇沉淀三次而将硫酸钠移除,获得具有四取代磺酸基的芳香化合物。以1h-nmr确认结构。其完全为辨识到原料、二磺化产物、三磺化产物,获得高纯度的四磺化产物。
对比例1和2都需要使用大量的强氧化性酸——发烟硫酸,反应温度较高(180-200℃)。这种条件下,对反应的容器及控制条件要求都很苛刻,造成成本高、不利于放大生产。
- 还没有人留言评论。精彩留言会获得点赞!