一种酰腙席夫碱铜配合物‑人血清白蛋白复合物及其应用的制作方法
本发明属于制药领域,具体涉及一种酰腙席夫碱铜配合物、一种酰腙席夫碱铜配合物-人血清白蛋白复合物及其在制备前药中的应用。
背景技术:
根据GLOBOCAN估计,2012年全球约有170万新乳腺癌病例和521,900例乳腺癌死亡,到2030年全球预计将增加2140万新癌症病例和约1320万癌症死亡病例。尽管传统癌症化疗在改善存活率方面在过去几十年是相当有益的,但它也显示出许多不利影响,包括有限的效率,无特异性,多药耐药性和不良副作用。为了克服这些障碍,已经开发了各种方法,包括前药策略和纳米颗粒药物递送策略。
目前,由于金属配合物具有广谱的几何形状和配位数以及动力学性质,将金属化合物开发为抗癌药物日益成为药物化学家的兴趣。在下一代金属基抗癌化合物中,Cu作为人体的必需元素,具有特定的生物活性和氧化性质,因此Cu抗癌化合物具有巨大的潜力。特别地,癌细胞比正常细胞摄取更多的Cu。目前设计HSA-金属基前药的主要方法是通过HSA上特殊的氨基酸与金属基药物化学偶联。但是,这种常规的化学偶联方法有诸多的缺点。例如,过多的化学链接剂会导致HSA沉淀。而Cys34处在HSA的深沟中不易连接药物。此外,在体内HSA载体运载金属前药到达癌细胞还存在两个可能的弊端。第一、金属前药连接在HSA表面,导致药物会暴露于体内的其他蛋白,以至于这些蛋白可能与HSA竞争结合药物,导致金属前药从HSA载体上逃逸而结合到其他蛋白可能导致副作用。第二、由于金属前药与HSA结合太弱,金属前药可能过早释放而不能到达癌细胞。
技术实现要素:
本发明针对现有技术不足,合成了两种具有离去基团的Cu配合物,该类配合物可以不使用任何化学连接剂共价结合在HSA的IIA结合区域,避免现有技术中存在的问题。进一步的,形成的酰腙席夫碱铜配合物-人血清白蛋白复合物可以通过去溶剂法制成人血清白蛋白铜(II)前药纳米颗粒(HSA-Cu(II)),药物均匀分散在纳米球体中,避免前药突释。
本发明具体技术方案如下:
一种酰腙席夫碱铜配合物,化学式如下:
[CuBr(BA)(L)]或者[CuBr(DMF)(L)],其中BA=喹啉,L为具有三齿酰腙席夫碱结构的化合物,DMF=N,N-二甲基甲酰胺),所述配合物中的Br-,喹啉或DMF作为配体与铜中心配位。
优选的,所述L为芳酰基腙席夫碱结构的化合物,更优选O,N,O–三齿芳酰基腙席夫碱结构的化合物。本发明的一个优选方案,L具有如下通式结构:
或者
其中
或-CH3,
R2=-H、-F、-Cl、-Br或-CH3。
本发明的一个优选方案,L为(E)-N'-(5-氯-2-羟基亚苄基)苯甲酰肼。所述酰腙席夫碱铜配合物优选如下:
本发明的另一目的在于提供上述酰腙席夫碱铜配合物与人血清白蛋白的复合物,上述酰腙席夫碱铜配合物可以不需要化学连接剂,而直接与人血清白蛋白共价结合形成。X-射线单晶结构研究表明HSA IIA结合区域的Lys199和His242氨基酸分别取代C1配合物中的喹啉和Br-配体与铜中心配位;而C2配合物的DMF基团保持惰性,HSA IIA结合区域的His242氨基酸取代轴向的Br-配体与铜中心配位。
上述酰腙席夫碱铜配合物-人血清白蛋白复合可以不使用化学连接剂,将酰腙席夫碱铜配合物与人血清白蛋白在室温下直接孵育24h制备得到,具体步骤如下:
(1)取2g粉末活性炭,5ml超纯水和10ml 20%的医用人血清白蛋白于50ml烧杯中,将该烧杯置于冰水中,调节pH到2.8,搅拌2h,然后调节pH到7.5。然后离心和透析去除除活性炭、HSA中的二聚体和多聚体获得后续试验所用的单体HAS;
(2)用紫外分光光度法测定透析后获得的单体HSA的浓度;
(3)以1:3的比例在20mM磷酸钾(pH 7.5)的缓冲溶液中混合HSA(100mg/mL)和铜配合物,然后常温孵育24h,过量的铜配合物通过离心洗脱去除。
本发明的另一目的在于提供上述酰腙席夫碱铜配合物-人血清白蛋白复合物纳米颗粒,可以将酰腙席夫碱铜配合物-人血清白蛋白复合物通过去溶剂技术制成大小均一且药物均匀的分散在纳米颗粒中的前药纳米球。
为了进一步提高靶向性和降低副作用,上述酰腙席夫碱铜配合物-人血清白蛋白复合物纳米颗粒可以进一步接上叶酸(FA)靶向小分子。
本发明的另一目的在于提供本发明所述的酰腙席夫碱铜配合物、酰腙席夫碱铜配合物-人血清白蛋白复合物、酰腙席夫碱铜配合物-人血清白蛋白复合物纳米颗粒以及连接有叶酸的酰腙席夫碱铜配合物-人血清白蛋白复合物纳米颗粒在制备前药中的应用。
上述前药为抗肿瘤药物或治疗铁过量疾病药物。
优选的,所述抗肿瘤药物可用于治疗宫颈癌,肺癌,乳腺癌,肝癌等癌症。
本发明优点:
(1)苯酰肼类席夫碱配体具有良好的抗癌活性,本发明保持主药效团配体(L)不变,设计具有不同配位原子的第二配体(喹啉或DMF),优选Br作为第三配体作为离去基团,在不使用化学连接剂的情况下,HSA的His242或者Lys199氨基酸可以取代本发明所述酰腙席夫碱铜配合物的离去基团而与铜中心配位,紧密结合到HSA的IIA子域,提高了HSA运载铜前药的效率。
(2)本发明所述酰腙席夫碱铜配合物-人血清白蛋白复合物纳米颗粒(HSA-C1/C2 NPs)及酰腙席夫碱铜配合物-人血清白蛋白复合物-叶酸纳米颗粒(FA-HSA-C1/C2 NPs)分别具有约162nm的直径和约220nm直径,两者都具有窄的粒度分布,注射后不容易被吞噬,而且具有良好的生物相容性。
(3)本发明所述酰腙席夫碱铜配合物(C1/C2)相比主药效团配体L具有更高抗肿瘤活性。
(4)本发明所述酰腙席夫碱铜配合物-人血清白蛋白复合物纳米颗粒(HSA-C1/C2 NPs)以及连接有叶酸的酰腙席夫碱铜配合物-人血清白蛋白复合物纳米颗粒(FA-HSA-C1/C2 NPs)相比与原型药物C1/C2具有更高抗肿瘤活性。
(5)本发明所述酰腙席夫碱铜配合物-人血清白蛋白复合物纳米颗粒(HSA-C1/C2 NPs)以及连接有叶酸的酰腙席夫碱铜配合物-人血清白蛋白复合物纳米颗粒(FA-HSA-C1/C2 NPs)相比与原型药物C1/C2对正常细胞具有更低的毒性。
(6)本发明所述酰腙席夫碱铜配合物-人血清白蛋白复合物纳米颗粒(HSA-C1/C2 NPs)以及连接有叶酸的酰腙席夫碱铜配合物-人血清白蛋白复合物纳米颗粒(FA-HSA-C1/C2 NPs)能明显降低酰腙席夫碱铜配合物的肝、肾毒性和良好的耐受性,具有很好的临床应用前景。
附图说明
图1.本发明所述酰腙席夫碱铜配合物[CuBr(BA)(L)](配合物C1)和[CuBr(DMF)(L)](配合物C2)的化学结构。
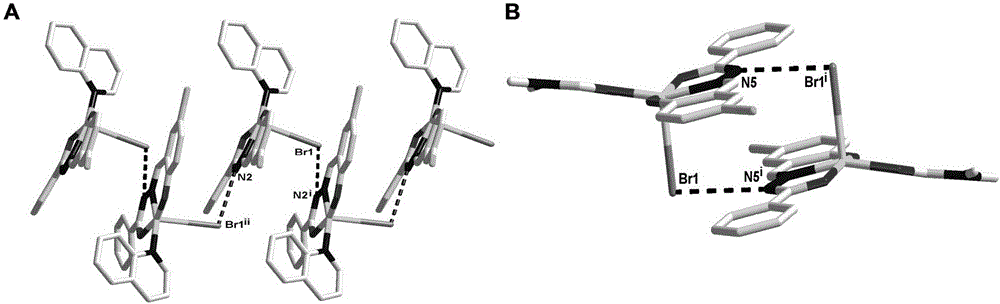
图2.配合物C1和配合物C2的N-H···Br的作用结果。(A)为配合物C1的N-H···Br作用形成的聚合键(对称代码:(i)x,0.5-y,-0.5+z);(B)为配合物C2的N-H···Br作用形成的二聚体(对称代码:(i)1-x,1-y,1-z)。
图3.(A)配合物C1和C2不同浓度下HSA的荧光猝灭光谱,T=298K,箭头表示在增加复合物浓度(0-4μM)时的荧光猝灭。(B)HSA-C1/C2复合物的MALDI-TOF-MS质谱图。
图4.HSA-C1复合物(A)和HSA-C2复合物(B)释放产物的电喷雾电离质谱(ESI-MS),黑细线是观察到的谱,直方灰线为模拟的m/z信号峰。
图5.(A)HSA-C1复合物和HSA-C2的2Fo-Fc电子密度图。(B)HSA-C1/C2复合物的整体结构。(C)铜配合物在HSA IIA结合区域的局部结合环境图。
图6.(A)HSA-C1/C2 NPs和FA-HSA-C1/C2 NPs的扫描电子显微镜图像(B)HSA-C1/C2 NPs和FA-HSA-C1/C2 NPs的粒径及分布。
图7.(A)C1/C2(5μM),HSA-C1/C2 NPs(5μM)和FA-HSA-C1/C2 NPs(5μM)作用于红细胞1h后的溶血反应分析。(B)相显微镜观察C1/C2(5μM),HSA-C1/C2 NPs(5μM)和FA-HSA-C1/C2 NPs(5μM)作用于红细胞的h后的凝集情况比较。每个值表示平均值±SD(n=3)。
图8.(A)Bel-7402肿瘤小鼠模型各给药组的肿瘤体积比较。(B)Bel-7402肿瘤小鼠模型各给药组的瘤重平均值比较。(C)Bel-7402肿瘤小鼠模型各给药组的体重比较,(*P<0.05;**P<0.01;***P<0.001)。
图9.体内活性实验各治疗组的肿瘤及其它器官切片的苏木精和伊红染色(H&E)组织学检查结果(放大倍数×400)。
图10.C2配合物、HSA-C2 NPs和FA-HSA-C2 NPs各治疗组地肿瘤组织和主要器官中铜含量(每个值表示平均值±SD(n=3):*P<0.05;**P<0.01.)。
具体实施方式
以下通过实施例说明本发明的具体步骤,但不受实施例限制。
在本发明中所使用的术语,除非另有说明,一般具有本领域普通技术人员通常理解的含义。
下面结合具体实施例并参照数据进一步详细描述本发明。应理解,该实施例只是为了举例说明本发明,而非以任何方式限制本发明的范围。
在以下实施例中,未详细描述的各种过程和方法是本领域中公知的常规方法。
下面结合具体实施例对本发明进一步说明。
医用人血清白蛋白从美国杰特贝林生物制品有限公司购买。所使用的其它化学药品和溶剂均为商业来源且是高纯度。在反应中所用的水全为蒸馏水。元素分析(C,N和H)在Perkin-Elmer公司2400分析仪上进行。红外(IR)测定使用Nexus870 FT-IR分光光度计(4000-400-1)和KBr压片。
实施例1本发明所述酰腙席夫碱铜配合物[CuBr(BA)(L)](化合物C1)和[CuBr(DMF)(L)](化合物C2)的制备和表征
本发明所用的酰腙席夫碱配体L以(E)-N'-(5-氯-2-羟基亚苄基)苯甲酰肼席夫碱(可参考文献:Ali HM,Puvaneswary S,Ng SW.5-Chlorosalicylaldehyde benzoylhydrazone.Acta Crystallogr.,Sect.E:Struct.Rep.Online 2005,61,o2415.制得)为例,制备酰腙席夫碱铜配合物。
[CuBr(BA)(L)](C1)的合成方法如下:L(0.55g,2mmol)和杂环氮配体喹啉(BQAL;0.26g,2mmol)溶解在15mL甲醇中,在常温搅拌1h,得到橙黄色液体。然后加入溶于甲醇的CuBr2(0.44g,2mmol)溶液。上述混合溶液继续在室温下再搅拌1h,得到墨绿色溶液,然后过滤混合溶液,收集滤液。该滤液保持在空气中缓慢挥发7天左右,溶液中慢慢析出蓝黑色晶体。将晶体分离,用蒸馏水洗涤三次,并在含有无水氯化钙的真空干燥器中干燥。产率:C23H17BrClCuN3O2(546.30)776mg(71%)。元素分析:C,50.56(50.53);H,3.13(3.14)和N,7.69(7.70)。IR(KBr,cm-1):1616ν(C=N);581,549,487,468,443ν(Cu-N/Cu-O)。
可参照上述方法,将DMF代替上述方法中的喹啉,制备[CuBr(DMF)(L)](C2)。产率:C17H17BrClCuN3O3(490.24)764mg(78%)。元素分析:C,41.65(41.53);H,3.49(3.52)和N,8.57(8.58)。IR(KBr,cm-1):1619ν(C=N);548,505,458,436,422ν(Cu-N/Cu-O)。
反应式如下:
单晶结构表征:选取形状规则无裂痕的单个晶体,在292-298K测定。用石墨单色器的Mo-Kα(ω-2θ的扫描方式)的辐射射线收集晶体数据,然后采用Bruker APEX SMART CCD面探衍射仪记录衍射数据。使用SADABS程序进行数据校正。结构通过重原子法和直接法解析,并使用SHELXTL5.1的全矩阵最小二乘方法对F2进行精制。所有的非氢原子进行各向异性精修。而所有的氢原子被放置在理想的几何位置并约束在它所属的原子上。化合物C1和C2的晶体学数据如表1所示。选择的部分键长和键角在如表2所示。用于结构分析的晶体学数据已被剑桥晶体学数据中心(Cambridge Crystallographic Data Centre)收集,C1的CCDC号为1011410,C2的CCDC号为967408。相应的晶体数据也可以免费从剑桥晶体数据中心获得。
表1.配合物C1和C2的晶体数据.
表2.配合物C1和C2的部分键长和键角[°]。
X-射线单晶衍射显示,配合物C1属于单斜晶系,空间群为P21/c。如图1所示,在不对称单元中含有一个三齿席夫碱配体,一个Cu(II)中心,一个喹啉配体和一个终端Br原子。Cu(II)中心与席夫碱配体上的一个氮原子和两个氧原子,一个来自喹啉配体的氮原子以及轴向终端Br配位,形成铜化合物常见的五配位结构。所有的Cu–N/O距离在的范围里,与已报道的相似结构键长一致。围绕Cu1中心配位多面体可以描述为一个略微扭曲的正方形棱锥(τ=0.004)。在固体状态下,C1的单体单元通过配位的溴原子(Br1)和邻近单体席夫碱配体上的N2i构成N-H···Br弱相互作用(Br1···N1i-H4i角度为158.7°;对称代码:(i)x,0.5-y,-0.5+z,如图2A所示),形成1D聚合连。
配合物C2的晶体结构如图1所示。X-射线单晶衍射显示,配合物C2属于三斜晶系,空间群为P-1。围绕铜中心的配位构型与C1十分相似,除了在平面上的配位分子DMF不一样。而在固体状态下,C2的单体单元通过配位的溴原子(Br1)和邻近单体席夫碱配体上的N5i构成N-H···Br弱相互作用(对称代码:(i)1-x,1-y,1-z,如图2B所示),形成二聚体结构。
实施例2酰腙席夫碱铜配合物-人血清白蛋白复合物(HSA-C1/C2)的制备和表征
取10ml的20%的医用人血清白蛋白、2g粉末活性炭和5ml超纯水于50ml烧杯中,将该烧杯置于冰水中,调节pH到2.8,搅拌2h,然后调节pH到7.5。然后离心和透析去除除活性炭、HSA中的二聚体和多聚体获得后续试验所用的单体HSA。
HSA复合物的单晶衍射:用20mM磷酸钾(pH 7.5)稀释脂肪酸(PA)到2.5mM。在试验中,为使C1和C2与HSA结合成前药复合物,先将380μL的PA(2.5mmol)、100μL的HSA(100mg/mL)、和90μL的Cu配合物(5mmol)混合,然后常温孵育24h,最后将混合物放在浓缩管中加水洗涤过滤浓缩至100mg/ml(微孔自旋过滤(10,000道尔顿截止)。通过在室温下坐滴蒸气扩散进行结晶,将等体积的HSA的复合物与贮存在座滴板中的溶液混合,此溶液由28-32%(重量/体积)聚乙二醇3350、50mM磷酸钾(pH 7.5)、5%甘油和5%的DMSO混合而成。长好的晶体直接从滴液中选择取出并然后在液氮中冷藏。X射线衍射的数据是使用上海同步辐射装置,在低温条件下(100K)收集完成的。衍射数据用HKL2000处理。HSA复合物的结构通过AMORE程序的分子置法(HSA-MYR为模型,PDB代码1BJ5)构建模型。构建的初模型首先用CNS程序刚性精修,接着增加精修轮数精修B-因子,直到获得原始的Fo-Fc和2Fo-Fc电子密度图。这些电子密度图被用来指示脂肪酸和铜化合物的位置,并通过手动调整结构和增加精修轮数进一步精修蛋白结构。PyMOL软件用于附图描绘和处理。表3中给出了数据收集的细节和晶胞参数。
表3.HSA-C1/C2复合物的晶体数据
a Values for the outermost resolution shell are given in parentheses.
bRmerge=100×ΣhΣj|Ihj-Ih|/ΣhΣj Ihj where Ih is the weighted mean intensity of the symmetry-related refractions Ihj.
cRmodel=100×Σhkl|Fobs-Fcalc|/ΣhklFobs where Fobs and Fcalc are the observed and calculated structure factors,respectively.
dRfree is the Rmodel calculated using a randomly selected 5%sample of reflection data omitted from the refinement.。
荧光光谱实验结果显示,随着配合物C1或C2浓度增加,人血清白蛋白在波长约346nm发生强烈的荧光淬灭,这表明这些铜配合物结合在HSA的IIA子域且有强烈结合能力(如图3A所示)。飞行质谱的基质辅助激光解吸/电离时间(MALDI-TOF-MS)显示HSA和C1或C2作用后,分子量比单独的HSA增加约400Da,表明约一个铜配合物结合到HSA(如图3B所示)。此外,在pH 5的条件下,从HSA-C1复合物中释放的铜化合物的电喷雾电离质谱(ESI-MS)表明,在源能量为0伏特时,存在m/z=335.97,这是[Cu(L)]+(fit:335.97)的同位素峰,这意味着Br和BA配体丢失(如图4A所示)。而在同样条件下,从HSA-C2复合物中释放的铜化合物的电喷雾电离质谱(ESI-MS)峰在m/z=409.03,这是[(L)Cu(DMF)]+(fit:409.03)的同位素峰,表明DMF配体存在于HSA-C2复合物中(如图4B所示)。
HSA-C1/C2复合物的单晶结构解析结果显示,在HAS的IIA子域有一个铜化合物分子(如图5A所示)。HSA-C1/C2的总体结构仍然是心脏形状。在IIA子域,铜化合结合到一个由Arg222,Arg218,His242,Lys199,Trp214,Leu219,Ala291,Phe223,Leu238,Arg257,Leu260,Ile264,Ser287和Ile290构成的疏水空腔中(如图5B所示)。HSA-C1结构显示,Lys199和His242分别取代喹啉配体和Br配体,与铜中心配位。而HSA-PA-C2结构显示,His242取代轴向Br配体,而Lys199没有取代平面上的DMF配体。
实施例3酰腙席夫碱铜配合物-人血清白蛋白复合物-叶酸纳米颗粒(FA-HSA-C1/C2)的制备和表征
(1)酰腙席夫碱铜配合物-人血清白蛋白复合物-叶酸纳米颗粒(HSA-C1/C2 NPs)的制备
为了获得HSA-金属前药和恒定载药比,首先将实施例1制得的本发明所述酰腙席夫碱铜配合物与HSA在室温下孵育(摩尔比1:3)24h,过量的酰腙席夫碱铜配合物离心透析去除。接着,按照文献中的去溶剂法,在室温和恒定搅拌的条件下,将4mL上述孵育的酰腙席夫碱铜配合物与HSA混合物(1.5mM)调节pH到8,连续加入15.0mL乙醇溶液(1mL/min),逐渐形成HSA-C1/C2 NPs。蛋白质变性后,加入8%的戊二醛水溶液450微升以实现纳米粒子交联。然后将上述混合物以40,000rpm离心10分钟,分离得到HSA-C1/C2 NPs。[Weber C,Kreuter J,Langer K.Desolvation process and surface characteristics of HSA-nanoparticles.Int J Pharm.2000;196(2):197-200;Sebak S,Mirzaei M,Malhotra M,Kulamarva A,Prakash S.Human serum albumin nanoparticles as an efficient noscapine drug delivery system for potential use in breast cancer:preparation and in vitro analysis.Int J Nanomedicine.2010;5:525-32.]。
(2)酰腙席夫碱铜配合物-人血清白蛋白复合物-叶酸纳米颗粒(FA-HSA-C1/C2 NPs)的制备
2.5mL的氢氧化钠(0.1N)FA溶液(20mg/mL),在50mg的N-(3-二甲基氨基丙基)-N-乙基碳二亚胺(EDC)和20mg的N-羟基琥珀酰亚胺(NHS)催化下避光搅拌30min。随后,将HSA-C1/C2 NPs混悬液(含量15mg/mL)加入,并继续避光搅拌3小时。未反应的FA通过差速离心(40,000rpm,10min)上述混合物去掉。将沉淀在底部的FA-HSA-C1/C2 NPs用水再分散,进行后续的实验。FA-HSA-C1/C2 NPs中铜化合物的含量通过裂解纳米颗粒成溶液状态,再用石墨炉原子吸收光谱仪(GF-AAS)测定其中铜元素的含量。
(3)FA-HSA-C1/C2 NPs的表征和溶血性测定
扫描电子显微镜图像显示,HSA-C1/C2 NPs和FA-HSA-C1/C2 NPs具有球形且光滑的表面(如图6A所示)。制备的纳米粒子的平均直径和粒度分布通过激光光散射测定。HSA-C1/C2 NPs和FA-HSA-C1/C2 NPs分别具有约162nm的直径和约220nm直径,且两者都具有窄的粒度分布(如图6B所示)。在这个粒径范围内,相比较大纳米颗粒,这种粒径的纳米颗粒注射后将不容易吞噬。
用于静脉给药的NPs必要条件之一是具有优良血液相容性。因此,对上述纳米颗粒的溶血率由溶血测定法进行评估。FA-HSA-C1/C2 NPs的溶血性性能通过分光光度法测定。对于红细胞凝集的研究,每个样品(5μM Cu(II)化合物,5μM HSA-C1/C2 NPs和5μM FA-HSA-C1/C2 NPs)与血液作用1h,置于在载玻片上,用相衬显微镜进行分析。结果图7A所示,相对于单独的C1/C2配合物,HSA-C1/C2 NPs和FA-HSA-C1/C2 NPs对红细胞溶血破坏大大降低,即使在5μM的高浓度。此外,单独的Cu(II)化合物与红细胞作用1小时,出现凝集现象,而在暴露于HSA-C1/C2 NPs和FA-HSA-C1/C2 NPs中红细胞没有观察到凝集现象(如图7B所示)。
实施例4 C1/C2、HSA-C1/C2 NPs以及FA-HSA-C1/C2 NPs的活性测定
(一)体外活性测定
肝癌细胞系Bel-7402和正常肺成纤维细胞WI-38(从the American Type Culture Collection和the German Collection of Microorganisms and Cell Cultures购买)在DMEM(具有的L-谷氨酰胺)培养基中培养。该培养基含有10%的胎牛血清(FBS),50U/mL的青霉素,50mg/mL链霉素。然后,将含有培养基的细胞置于37℃水汽湿度和5%CO2的培养箱培养。
将密度约为3×104细胞/mL,接种到96孔板中(每孔180μL)。在温度37℃和5%CO2的CO2培养箱中培养24h。然后加入具有不同浓度梯度的待测药品(C1/C2、HSA-C1/C2 NPs和FA-HSA-C1/C2 NPs,药品浓度以铜计),再在37℃和5%CO2的条件下孵育48h。吸光度值用酶标仪在570/630nm的双波长下测。最终的IC50(存活细胞数是对照组一半的时候,药物浓度)值由经典的Bliss方法得出(n=5)。所有测试实验至少3次独立的重复。
结果如表4所示,结果表明在体外,FA-HSA-C1/C2 NPs比C1/C2和HSA-C1/C2 NPs对叶酸受体分泌较多的Bel-7402细胞表现出更高的细胞毒活,而对叶酸受体分泌很少的正常WI-38肺成纤维细胞中没有观察到类似的效果。
表4 C1/C2、HSA-C1/C2 NPs和FA-HSA-C1/C2 NPs对Bel-7402和WI-38细胞的IC50a(μM)值。
aIC50 values are presented as the mean±SD from three separated experiments。
(二)体内活性测定
体外活性研究表明C2系列(C2,HSA-C2 NPs和FA-HSA-C2 NPs)比C1系列(C1,HSA-C1 NPs和FA-HSA-C1 NPs)有更好的抗癌活性。因此,为了进一步评估FA-官能化的HSA纳米载体和前药策略在体内是否表现出更好的抗癌性质,以活性较好的C2系列为例,采用异种移植Bel-7402鼠肝癌模型进行进一步研究。
所有实验动物均遵守中华人民共和国卫生管理条例(文件编号NO.55,2001)。Kunming(KM)小鼠由中国科学院(上海)提供。
所有实验重复3到5次。Student's t-test检验用于评估测量值的差异显著性。测量结果以平均值±标准差(SD)表示,当p<0.05,认为具有显著性差异。
(1)急性毒性研究
C2,HSA-C2 NPs和FA-HSA-C2 NPs在老鼠体内的急性毒性实验参考文献中的方法。32只健康的KM小鼠(年龄3~4周,重18~22g,等量的雌性和雄性),分成4组,对照组,C2组,HSA-C2 NPs组和FA-HSA-C2 NPs组,每组8只。C2组,HSA-C2 NPs和FA-HSA-C2 NPs组注射等量的药量(15μmol Cu/kg体重),对照组注射等体积的0.9%生理盐水。作用7天后,分析血液样本中的天(门)冬氨酸转氨酶(aspartate aminotransferase,AST),丙氨酸转氨酶(alanine aminotransferase,ALT),肌酸激酶(creatinine kinase,CK)和血尿氮(blood urea nitrogen,BUN)指标。
通过测定加药7天后,天(门)冬氨酸转氨酶(aspartate aminotransferase,AST),丙氨酸转氨酶(alanine aminotransferase,ALT),肌酸激酶(creatinine kinase,CK)和血尿氮(blood urea nitrogen,BUN)水平,评估对正常老鼠的心、肝和肾脏的毒性(如表5所示)。所有加药组CK值水平与对照组相似,表明C2,HSA-C2 NPs和FA-HSA-C2 NPs几乎没有心脏毒性。高的BUN值对应高的肾脏毒性。C2组的BUN值(18.4±2.4mmol/L)明显高于对照组(NaCl,7.3±1.1mmol/L)。而HSA-C2 NPs和FA-HSA-C2 NPs组,特别是FA-HSA-C2 NPs组,明显有较低的肾毒性。C2组的AST和ALT水平明显高于对照组,而HSA-C2 NPs和FA-HSA-C2 NPs组的AST和ALT水平与对照组相当,表明HSA-C2 NPs和FA-HSA-C2 NPs有很低的肝毒性。
表5.NaCl,C2,HSA-C2 NPs and FA-HSA-C2 NPs给药组小鼠的血清学分析结果
(2)体内抗肿瘤活性研究
将Bel-7402细胞和高浓度基质胶混匀(基质胶:PBS=1:3)。由于基质胶在10℃即凝结,因此,所有过程均应在冰上操作。调整细胞密度,每只裸鼠/0.2mL,为了保证每只裸鼠接种的细胞数均匀,要将0.2mL混匀的细胞分至预冷的EP管中,细胞数为5×106个,每只裸鼠接种一管。接种时所用的1mL注射器也应预冷。接种细胞后,观察和记录肿瘤的形状和大小。
当肿瘤长约100mm3,挑选瘤体均匀、大小相对一致的32只裸鼠随机分为4组,每组8只(对照组,C2组,HSA-C2 NPs组和FA-HSA-C2 NPs组)。对照组注射0.9%生理盐水,C2组,HSA-C2 NPs组和FA-HSA-C2 NPs组以静脉注射给药方式注射3.5μmol Cu/kg体重的药物。每3天给一次药,给药前称裸鼠体重并用游标卡尺记录肿瘤体积(肿瘤体积(mm3)=1/2×(L×W2),L为肿瘤的长径,W为肿瘤的短径),共给药14次。
Bel-7402肿瘤小鼠分别静脉注射C2,HSA-C2 NPs和FA-HSA-C2 NPs,NaCl注射作为对照。每3天监测一次肿瘤体积和老鼠体重的变化。24天实验期结束后,FA-HSA-C2 NPs治疗组的肿瘤明显小于C2和HSA-C2 NPs组(结果如图8A所示)。相比对照组,24天治疗后,C2组肿瘤体积为对照组体积的65.3±5.9%,HSA-C2 NPs是对照组的47.6±5.1%,FA-HSA-C2 NPs仅为对照组的29.9±3.1%,表明FA-HSA-C2 NPs比C2和HSA-C2 NPs组有更好抑瘤功效。同时,治疗后的24天,处死小鼠收获肿瘤、称重和拍照(如图8B和C所示)。肿瘤抑制率(TIR)用肿瘤重量来计算。相比对照组,FA-HSA-C2 NPs的TIR为78.2%(P<0.001),明显高于C2的肿瘤抑制率(34.2%,P<0.01)和HSA-C2 NPs的抑制率(54.4%,P<0.001)。
为了进一步评估抗癌活性,肿瘤组织进行切片和苏木精和伊红染色(H&E)组织学检查。H&E染色显示,药物组与对照组的组织切片的肿瘤组织形态学明显不一样(如图9所示)。在NaCl组中,肿瘤细胞中有一个纺锤形大核,并在肿瘤组织中没有可见的细胞凋亡或坏死,表示肿瘤快速生长。然而,每个显微镜视野中显示的肿瘤细胞,在C2,HSA-C2 NPs和FA-HSA-C2 NPs组中平均细胞数量下降,出现碎片和核的收缩,特别是FA-HSA-C2 NPs治疗的肿瘤更加明显。
(3)体内副作用比较
图8C显示了治疗过程中,对照组,C2,HSA-C2 NPs和FA-HSA-C2 NPs组老鼠体重的变化。HSA-C2 NPs和FA-HSA-C2 NPs组老鼠体重相对于初体重分别下降了11.8%和8.1%,明显低于对照组老鼠体重的下降(16.5%)。而C2组老鼠体重的下降达到18.4%。这些结果显示,HSA-C2 NPs和FA-HSA-C2 NPs降低了C2的副作用。此外,H&E染色也用于观察药物相关的副作用和对主要器官的毒性(如图9所示)。值得一提,所有组对心脏几乎没有影响。然而,可以清楚地观察到C2诱导严重的肝脏损害(肝细胞萎缩和轻度脂肪病变)和肾脏损害(焦点异常和肾小管的透明管型)。与此相反,HSA-C2 NPs和FA-HSA-C2 NPs对肝脏和肾脏损伤较小-轻微炎症和细胞萎缩。体重变化和病理研究结果表明,HSA-C2 NPs和FA-HSA-C2 NPs可以有效地减少C2诱导的副作用和毒性,特别是叶酸官能化的HSA-C2 NPs。
(4)体内靶向性研究
为了确定在体内叶酸官能化的HSA NPs载体能否有效的积累到Bel-7402肿瘤组织中,我们测定了C2,HSA-C2 NPs和FA-HSA-C2 NPs治疗组中肿瘤组织和主要器官中铜含量。将保存于-80℃冰箱的荷瘤鼠的心、肝、肾脏和肿瘤组织分别匀浆。然后分别取0.5g样品加入1mL 30%过氧化氢(H2O2)和7mL的浓HNO3,在聚四氟乙烯压力容器中矿化。矿化后的样品中的Cu离子含量用电感耦合等离子原子发射光谱(ICP-AES)测定。
电感耦合等离子体原子发射光谱法(ICP-AES)的结果表明,HSA-C2 NPs和FA-HSA-C2 NPs组肿瘤中铜的含量分别是C2组的1.5和2.1倍(如图10所示)。这些结论表明,叶酸受体介导的细胞内屯作用对叶酸官能化的HSA NPs载体选择性积累到Bel-7402肿瘤细胞起到重要的作用。结果数据还显示,HSA-C2 NPs和FA-HSA-C2 NPs有利于降低C2积累到肝组织和肾脏组织中。
- 还没有人留言评论。精彩留言会获得点赞!