ATG类似物及其制备方法和应用与流程
本发明涉及一种ATG类似物,本发明还涉及ATG类似物的制备方法,本发明还涉及上述ATG类似物的应用,属于医药领域。
背景技术:
牛蒡苷元ATG具有多种生物活性。然而,对于一种潜在的药物而言,即使具有显著的疗效,但是能否广泛的开展应用,以及在实际应用于人体上的效果究竟如何,仍然需要进一步的性质的改进。而天然的ATG具有水溶性低和易在一级代谢中被分解的特点,这限制了其进一步的应用。
技术实现要素:
本发明的目的在于提供一种ATG的类似物,以解决上述问题。
本发明采用了如下技术方案:
本发明提供下列三种通式化合物:
其中,X/Y/Z可以分别选自C、N、S、O。
R1,R2,R3,R4,R5各自分别选自:H,CH3基团,脂肪族或者芳香族碳氢化合物基团,硝基,磷酸基,硫化物基团,砜类基团,腈类基团,杂环基,硼酸酯或硼酸基团,酯类基团,胺类基团。
本发明还提供B部分为开环的化合物,结构式如下:
这些化合物均为ATG的类似物,具有与ATG相似的化学性质和生物活性。
本发明提供了上述ATG类似物作为PP2A激动剂的应用,提高了PP2A的调控效果。
优选的,效果最佳的三种化合物是BT280,BT281和BT282。
更优选的,效果更佳的是BT282。
本发明还提供了BT280,BT281和BT282的化学合成方法,以上列举的其它化合物也可以使用类似的方法进行合成。
首先合成化合物后面简称之为化合物C,然后用硼基反应合成BT280,再从BT280和水溶的3MKHF2中合成BT282。然后BT282进一步在二氧化硅和水的条件下被酸化,从而得到BT281硼酸衍生物。具体如下:
化合物C的合成.3-(4-bromophenyl)-5-(3,4,5-trimethoxyphenyl)-1,2,4-oxadiazole
化合物C的合成步骤:
三甲氧基苯甲酸A,用量1.0毫摩尔(0.212克);和B,用量1.2毫摩尔(0.195克)加催化剤CDI(N,N'-二环己基碳二亚胺),溶解在1.8毫升DMF中并在室温下搅拌30分钟合成XB-A-26(C)。加入XB-A-26,用量1.2毫摩尔(0.258克),并将反应混合物在回流下在80℃加热24小时,通过TLC监测。将混合物倾入20毫升水中并用EtOAc(3×10mL)萃取,将合并层用Na2SO4干燥,过滤并真空浓缩。将粗产物通过硅胶纯化(己烷:EtOAc=3:1),得到为白色固体的恶二唑产物,这样能得到0.30克的终产物C。
BT280的合成:3-(4-(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)苯基)-5-(3,4,5-三甲氧基苯基)-1,2,4-恶二唑。3-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl phenyl)-5-(3,4,5-trimethoxyphenyl)-1,2,4-oxadiazole
BT280的合成步骤:取化合物C(0.77毫摩尔,0.30g),B2Pin2(频那醇硼酸酯)(1.07毫摩尔,0.273克),ACOK(3.85毫摩尔,0.377克),Pd(PPh3)2Cl2(0.077毫摩尔,0.054克)和DMSO(15毫升)一起加入50mL圆底烧瓶中。将所得混合物在室温下搅拌10分钟,然后将所得混合物在80摄氏度下搅拌1天。用水(30毫升)稀释该混合物,用乙酸乙酯(3×30毫升)萃取。将有机层经硫酸钠干燥并真空浓缩。将所得进行纯化柱提取,可以得到0.06克纯品BT280。
BT282的合成:钾三氟(4-(5-(3,4,5-三甲氧基苯基)-1,2,4-恶二唑-3-基)苯基)硼酸盐
步骤:
将BT280(0.29毫摩尔,0.12克)加入到15毫升MeOH中,然后加入3M的KHF2,搅拌2小时后,蒸发MeOH得到残留。将该残留物在丙酮中溶解并过滤,该滤液蒸干后得到白色固体,进一步经过EtOAC洗脱得到65毫克最终纯品。
BT281的合成:(4-(5-(3,4,5-三甲氧基苯基)-1,2,4-恶二唑-3-基)苯基)硼酸
步骤:将BT282(37毫克,0.9毫摩尔)和硅胶(0.18毫摩尔,11毫克)加入到H2O/乙酸乙酯(1.0毫升)中。将反应混合物在室温下搅拌直至反应完全。将反应混合物过滤除去硅胶,将滤饼用丙酮洗涤。水相和有机分离,将有机层干燥得到目的产品BT281。
本发明还提供了上述ATG类似物,尤其是BT280,BT282和BT282在提高PP2A活性中的应用。
发明的有益效果
本发明的ATG类似物由于具有噁二唑环,因此其具备更多亲水性的特点并大幅度增加了水溶性。
在A部分,由于三甲氧基代替苯基环衍生物增加跨细胞膜疏水性特点,并且保护羟基避免终止于一级代谢。
在C部分,引入硼酸酯代替苯基环,提高了与生物分子结合的可能性。
BT280,BT81和BT282均能够提高PP2A的生物活性。其中BT282较天然ATG的效果更好。
附图说明
图1是本发明的几种ATG类似物对PP2A活性的影响。
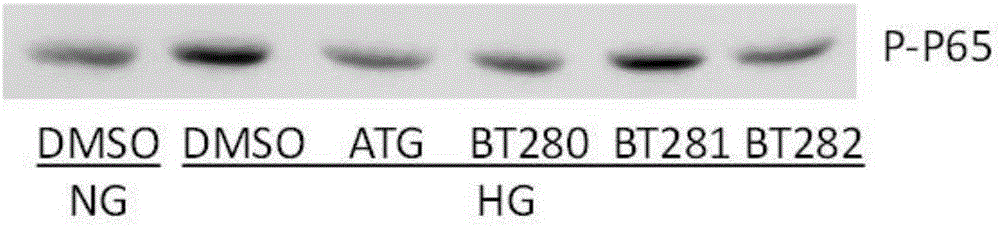
图2是本发明的几种ATG类似物对NF-kB磷酸化程度的影响。
具体实施方式
以下结合附图来说明本发明的具体实施方式。
本发明提供一系列ATG同型物:包括ABC三个部分,B部分有开环和闭环两种形式,在两种形式下,XYZ分别选自C、N、S、O。R1,R2,R3,R4,R5各自分别选自:H,CH3基团,脂肪族或者芳香族碳氢化合物基团,硝基,磷酸基,硫化物基团,砜类基团,腈类基团,杂环基,硼酸酯或硼酸基团,酯类基团,胺类基团。
进一步,上述类似物中还包括B部分为开环结构的化合物,如下:
上引入含硼的药效基团,内酯环改变为噁二唑环,使其具备更多亲水性的特点并尽可能增加水溶性。经过筛选,效果最佳的是BT280、BT81、BT282三个化合物。
(1)ATG类似物的结构特点:
含硼的药效基团,含硼的基团不仅可以与靶蛋白通过氢键结合也可以通过共价键结合从而产生生物活性,因此以ATG为目标确定了三种化合物:BT280,BT281,and BT282,具体结构见上面化学式。
合成的ATG同型剂结构如以上化学式所示。化合物的结构,原始的ATG包含三部分,A部分苯基环衍生物,B部分内酯环和苯基环系统。在B部分,将内酯环修正为噁二唑环,使其具备更多亲水性的特点并尽可能增加水溶性。在C部分,引入硼酸酯代替苯基环,含硼的基团不仅可以与靶蛋白通过氢键结合也可以通过共价键结合从而产生生物活性。在A部分,用三甲氧基代替苯基环衍生物增加跨细胞膜疏水性特点,并且保护羟基避免终止于一级代谢。合成了三种化合物,BT280,BT281和BT 282。
(2)ATG类似物的生物活性:
通过检测下游PP2A活性和NF-kB磷酸化程度来判断该三种化合物的生物活性,结果如图1所示,以DMSO为参照物。分别使用天然ATG和新化合物BT280、BT281和BT282分别刺激293T细胞,ATG和三种新化合物的浓度均选择10μM,可见BT280对PP2A活性的影响程度与天然ATG相似。而BT281对PP2A活性的提高作用较天然ATG弱一些,但仍然强于DMSO组。化合物BT282对PP2A的活性提高作用高于天然ATG。因此三种化合物均能够用于提高PP2A的生物活性。
显然BT282从提高PP2A的活性上来讲是最佳选择。BT280和BT281虽然在提高PP2A活性上与天然ATG相比并没有明显的提升,但是由于BT280和BT281具有更好的水溶性和更佳的生物反应基团,提示其在动物体级别的用药中,能够提供更好的给药效果,例如由于其较佳的水溶性,可以更好的通过动物体的循环系统到达患处,并达到更高的给药浓度,从而达到更佳的给药效果。
对于NF-kB磷酸化程度的结果可见图2。可见NF-kB磷酸化程度与图1中的PP2A结果相对应。提示ATG的类似物在NF-kB通路中的作用也与ATG相似。显示ATG类似物也可以用于对NF-kB的磷酸化程度进行调节。图2的结果中,ATG类似物的刺激浓度为10μM。
综上所述,BT282是最佳的目标化合物。BT282与ATG相比提升的最明显。但是其它两个化合物也可用,相对于天然ATG,BT280和BT281虽然活化PP2A的效果没有优势,但是在具体的生物体实验中,由于在水溶性上较天然ATG更具优势,因此BT280和BT281仍然可以作为潜在的药物对象。
合成方法:
<实施例一>
首先合成化合物后面简称之为化合物C,然后用硼基反应合成BT280,再从BT280和水溶的3MKHF2中合成BT282(三氟钾盐)。然后该三氟钾盐复合物进一步在二氧化硅和水的条件下被酸化,从而得到BT281硼酸衍生物。
化合物C的合成.3-(4-bromophenyl)-5-(3,4,5-trimethoxyphenyl)-1,2,4-oxadiazole
化合物C的合成步骤:
化合物A为三甲氧基苯甲酸,用量0.9毫摩尔;和化合物B,用量1.1毫摩尔加催化剤CDI(N,N'-二环己基碳二亚胺),溶解在1.8毫升DMF中并在室温下搅拌30分钟合成XB-A-26(C)。加入XB-A-26,用量1.4毫摩尔,并将反应混合物在回流下在75℃加热36小时,通过TLC监测。将混合物倾入20毫升水中并用EtOAc(3×10mL)萃取,将合并层用Na2SO4干燥,过滤并真空浓缩。将粗产物通过硅胶纯化(己烷:EtOAc=3:1),得到为白色固体的恶二唑产物,这样能得到0.25克的终产物C。
BT280的合成:3-(4-(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)苯基)-5-(3,4,5-三甲氧基苯基)-1,2,4-恶二唑。3-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl phenyl)-5-(3,4,5-trimethoxyphenyl)-1,2,4-oxadiazole
BT280的合成步骤:取化合物C(0.75毫摩尔),B2Pin2(频那醇硼酸酯)(1.05毫摩尔),ACOK(3.80毫摩尔),Pd(PPh3)2Cl2(0.075毫摩尔)和DMSO(15毫升)一起加入50mL圆底烧瓶中。将所得混合物在室温下搅拌10分钟,然后将所得混合物在85摄氏度下搅拌1天。用水(30毫升)稀释该混合物,用乙酸乙酯(3×30毫升)萃取。将有机层经硫酸钠干燥并真空浓缩。将所得进行纯化柱提取,可以得到0.05克纯品BT280。
BT282的合成:钾三氟(4-(5-(3,4,5-三甲氧基苯基)-1,2,4-恶二唑-3-基)苯基)硼酸盐
步骤:
将BT280(0.28毫摩尔)加入到15毫升MeOH中,然后加入3M的KHF2,搅拌2小时后,蒸发MeOH得到残留。将该残留物在丙酮中溶解并过滤,该滤液蒸干后得到白色固体,进一步经过EtOAC洗脱得到纯品,62毫克最终纯品。
BT281的合成:(4-(5-(3,4,5-三甲氧基苯基)-1,2,4-恶二唑-3-基)苯基)硼酸
步骤:将BT282(36毫克)和硅胶(0.18毫摩尔)加入到H2O/乙酸乙酯(1.0毫升)中。将反应混合物在室温下搅拌直至反应完全。将反应混合物过滤除去硅胶,将滤饼用丙酮洗涤。水相和有机分离,将有机层干燥得到目的产品BT281。
<实施例二>
首先合成化合物后面简称之为化合物C,然后用硼基反应合成BT280,再从BT280和水溶的3MKHF2中合成BT282(三氟钾盐)。然后该三氟钾盐复合物进一步在二氧化硅和水的条件下被酸化,从而得到BT281硼酸衍生物。
化合物C的合成.3-(4-bromophenyl)-5-(3,4,5-trimethoxyphenyl)-1,2,4-oxadiazole
化合物C的合成步骤:
化合物A:三甲氧基苯甲酸,用量1.0毫摩尔;和化合物B,用量1.2毫摩尔加催化剤CDI(N,N'-二环己基碳二亚胺),溶解在1.8毫升DMF中并在室温下搅拌30分钟合成XB-A-26(C)。加入XB-A-26,用量1.3毫摩尔,并将反应混合物在回流下在80℃加热24小时,通过TLC监测。将混合物倾入20毫升水中并用EtOAc(3×10mL)萃取,将合并层用Na2SO4干燥,过滤并真空浓缩。将粗产物通过硅胶纯化(己烷:EtOAc=3:1),得到为白色固体的恶二唑产物,这样能得到0.30克的终产物C。
BT280的合成:3-(4-(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)苯基)-5-(3,4,5-三甲氧基苯基)-1,2,4-恶二唑。3-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl phenyl)-5-(3,4,5-trimethoxyphenyl)-1,2,4-oxadiazole
BT280的合成步骤:取化合物C(0.77毫摩尔,0.30g),B2Pin2(频那醇硼酸酯)(1.07毫摩尔,0.273克),ACOK(3.85毫摩尔,0.377克),Pd(PPh3)2Cl2(0.077毫摩尔,0.054克)和DMSO(15毫升)一起加入50mL圆底烧瓶中。将所得混合物在室温下搅拌10分钟,然后将所得混合物在80摄氏度下搅拌1天。用水(30毫升)稀释该混合物,用乙酸乙酯(3×30毫升)萃取。将有机层经硫酸钠干燥并真空浓缩。将所得进行纯化柱提取,可以得到0.06克纯品BT280。
BT282的合成:钾三氟(4-(5-(3,4,5-三甲氧基苯基)-1,2,4-恶二唑-3-基)苯基)硼酸盐
步骤:
将BT280(0.29毫摩尔,0.12克)加入到15毫升MeOH中,然后加入3M的KHF 2,搅拌2小时后,蒸发MeOH得到残留。将该残留物在丙酮中溶解并过滤,该滤液蒸干后得到白色固体,进一步经过EtOAC洗脱得到纯品,65毫克最终纯品。
BT281的合成:(4-(5-(3,4,5-三甲氧基苯基)-1,2,4-恶二唑-3-基)苯基)硼酸
步骤:将BT282(37毫克,0.9毫摩尔)和硅胶(0.18毫摩尔,11毫克)加入到H2O/乙酸乙酯(1.0毫升)中。将反应混合物在室温下搅拌直至反应完全。将反应混合物过滤除去硅胶,将滤饼用丙酮洗涤。水相和有机分离,将有机层干燥得到目的产品BT281。
<实施例三>
首先合成化合物后面简称之为化合物C,然后用硼基反应合成BT280,再从BT280和水溶的3MKHF2中合成BT282(三氟钾盐)。然后该三氟钾盐复合物进一步在二氧化硅和水的条件下被酸化,从而得到BT281。
化合物C的合成.3-(4-bromophenyl)-5-(3,4,5-trimethoxyphenyl)-1,2,4-oxadiazole
化合物C的合成步骤:
化合物A:三甲氧基苯甲酸,用量1.1毫摩尔;和化合物B,用量1.3毫摩尔加催化剤CDI(N,N'-二环己基碳二亚胺),溶解在1.8毫升DMF中并在室温下搅拌30分钟合成XB-A-26(C)。加入XB-A-26,用量1.3毫摩尔,并将反应混合物在回流下在85℃加热20小时,通过TLC监测。将混合物倾入20毫升水中并用EtOAc(3×10mL)萃取,将合并层用Na2SO4干燥,过滤并真空浓缩。将粗产物通过硅胶纯化(己烷:EtOAc=3:1),得到为白色固体的恶二唑产物,这样能得到0.31克的终产物C。
BT280的合成:3-(4-(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)苯基)-5-(3,4,5-三甲氧基苯基)-1,2,4-恶二唑。3-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl phenyl)-5-(3,4,5-trimethoxyphenyl)-1,2,4-oxadiazole
BT280的合成步骤:取化合物C,0.79毫摩尔,B2Pin2(频那醇硼酸酯)1.08毫摩尔,ACOK,3.88毫摩尔,Pd(PPh3)2Cl2,0.079毫摩尔,和DMSO,15毫升。一起加入50mL圆底烧瓶中。将所得混合物在室温下搅拌10分钟,然后将所得混合物在80摄氏度下搅拌1天。用水(30毫升)稀释该混合物,用乙酸乙酯(3×30毫升)萃取。将有机层经硫酸钠干燥并真空浓缩。将所得进行纯化柱提取,可以得到0.07克纯品BT280。
BT282的合成:钾三氟(4-(5-(3,4,5-三甲氧基苯基)-1,2,4-恶二唑-3-基)苯基)硼酸盐
步骤:
将BT280,0.32毫摩尔,加入到15毫升MeOH中,然后加入3M的KHF 2,搅拌2小时后,蒸发MeOH得到残留。将该残留物在丙酮中溶解并过滤,该滤液蒸干后得到白色固体,进一步经过EtOAC洗脱得到纯品,66毫克最终纯品。
BT281的合成:(4-(5-(3,4,5-三甲氧基苯基)-1,2,4-恶二唑-3-基)苯基)硼酸
步骤:将BT282,39毫克和硅胶(0.18毫摩尔,11毫克)加入到H2O/乙酸乙酯(1.0毫升)中。将反应混合物在室温下搅拌直至反应完全。将反应混合物过滤除去硅胶,将滤饼用丙酮洗涤。水相和有机分离,将有机层干燥得到目的产品BT281。
- 还没有人留言评论。精彩留言会获得点赞!