一种马来酰亚胺基团封端型的苯并噁嗪齐聚物及其制备方法与流程
本发明属于热固性树脂及其制备技术领域,具体的说明涉及一种马来酰亚胺基团封端型的苯并噁嗪齐聚物及其制备方法。
技术背景
苯并噁嗪树脂以其与传统酚醛树脂相当的力学性能,电性能以及耐高温,耐烧蚀性能而受到广泛关注,不仅如此,还改进了酚醛树脂的不足之处,如脆性大,固化时有小分子释出,强酸催化腐蚀设备等。苯并噁嗪一般由酚类化合物,胺类化合物和甲醛经缩合反应制得,在加热或催化剂作用下可开环聚合,得到含氮且类似酚醛树脂的网状结构的聚合物。苯并噁嗪树脂虽然具有上述优点,但也因为固化温度偏高而造成了加工成型的困难,限制了其在某些方面的应用。
基于其上述优点,苯并噁嗪树脂在国内外的研究工作都十分活跃。这些工作基于苯并噁嗪灵活多变的分子设计性,采用不同的酚源和胺源合成出了性能不同的苯并噁嗪,从而满足不同的应用场合。如中国发明专利申请《含N-烯丙基的苯并噁嗪中间体和组合物及其制备方法(公开号为CN 1472205A)》提供了含N-烯丙基的苯并噁嗪中间体的制备方法,产物具有优良的耐高温性能、力学性能、阻燃性能和加工性能。中国发明专利申请公开《一种含马来酰亚胺和烯丙基醚的苯并噁嗪及其制备方法(公开号:CN 11235033A)》,申请是将现有对位马来酰亚胺基团基础上引入烯丙基醚,来改善苯并噁嗪树脂的性能。中国发明专利申请《含酰亚胺结构的双苯并噁嗪及其制备法(公开号为CN 101857592 A)》公开了含酰亚胺结构的双苯并噁嗪及其制备方法,改善了聚合物的耐热性和力学性能。但到目前为止,还没有报道涉及到马来酰亚胺基团封端的主链型苯并噁嗪的合成及应用。
本发明在苯并噁嗪主链上引入马来酰亚胺基团,缩短了苯并噁嗪的合成时间,同时,除了噁嗪环开环固化交联外,马来酰亚胺基团还可以进一步双交联反应,得到一种更为致密的双交联苯并噁嗪树脂。大大提高了树脂的热稳定性,还改善了其加工性,有利于后续加工成型。本发明将进一步拓宽苯并噁嗪树脂的应用领域。
技术实现要素:
本发明针对苯并噁嗪树脂现存的缺陷,通过苯并噁嗪化学引入可交联的马来酰亚胺基团,简化苯并噁嗪的合成工艺,而引入的马来酰亚胺基团减弱了分子的刚性,提高了树脂的可加工性,并且可通过端基多重交联显著提高树脂的热、力学性能。
本发明提供的一种马来酰亚胺基团封端型的苯并噁嗪齐聚物,其分子式如下:
所述马来酰亚胺基团在苯并噁嗪环氧原子的邻位、间位或对位。
其中,-R1-为下列之一:
-R2-为下列之一:
所述苯并噁嗪齐聚物经进一步固化交联后,其聚苯并噁嗪树脂的玻璃化转变温度为250-400℃。
一种马来酰亚胺基团封端型的苯并噁嗪齐聚物的制备方法,包括以下步骤:
(1)马来酰亚胺官能化酚的合成:
将氨基苯酚和马来酸酐按适当摩尔比混合,加入到N,N-二甲基甲酰胺(DMF)为溶剂的冰浴体系中,之后向其中加入五氧化二磷以及浓度为95-98%的浓硫酸,将该体系转移到油浴锅中,然后将反应体系中充满氮气,反应温度为70-120℃,反应6-8小时,停止反应后,将反应液经去离子水水洗、过滤、干燥,得到马来酰亚胺官能化酚;
反应方程式如下:
(2)含马来酰亚胺基团的的苯并噁嗪齐聚物的合成:
将步骤(1)制得的马来酰亚胺官能化酚、二胺类化合物、二酚类化合物和多聚甲醛按适当摩尔比混合,加入低极性溶剂中,然后加热升温,使反应体系在120℃反应5h,停止反应后,将反应液用碱液A洗涤2-3次,悬蒸溶剂得到沉淀,得到产物;
反应方程式如下:
其中,-R1-为下列之一:
-R2-为下列之一:
步骤(1)中,所述的氨基苯酚与马来酸酐按摩尔比为1:1~1:1.5;优选比例为1:1.1。
步骤(1)中,所述的五氧化二磷的质量占氨基苯酚与马来酸酐总质量的20~40%,95-98%的浓硫酸占氨基苯酚与马来酸酐总质量的10~20%。P2O5为除水剂、H2SO4为催化剂。
步骤(2)中,所述的马来酰亚胺官能化酚、二胺类化合物、二酚类化合物和多聚甲醛按摩尔比为2:n:(n+1):4(n+1),其中,1≤n≤20,且为整数。
步骤(2)中,所述的低极性溶剂为甲苯或二甲苯。
步骤(2)中,所述的碱液A为氢氧化钠、氢氧化钾、碳酸钠、碳酸氢钠、碳酸钾或碳酸氢钾中的一种。
本发明所得产物的结构经核磁共振、红外光谱、元素分析等分析方法得到证实。根据本发明合成的马来酰亚胺基团封端的苯并噁嗪齐聚物,玻璃化转变温度Tg高达250~400℃。
本发明制得的马来酰亚胺封端型苯并噁嗪齐聚物可作为基体树脂应用于纤维增强材料、刹车片及其航空航天领域。
本发明的有益效果为:
通过苯并噁嗪化学引入可交联的马来酰亚胺基团,缩短反应时间并简化工艺,而引入的马来酰亚胺基团在固化时可二次交联,进一步提高了树脂的交联度,提高了树脂的热、力学性能。此外,此方法制备工艺简单,对设备要求低,可批量化生产。
附图说明
图1实施例1得到的苯并噁嗪齐聚物的红外光谱图;
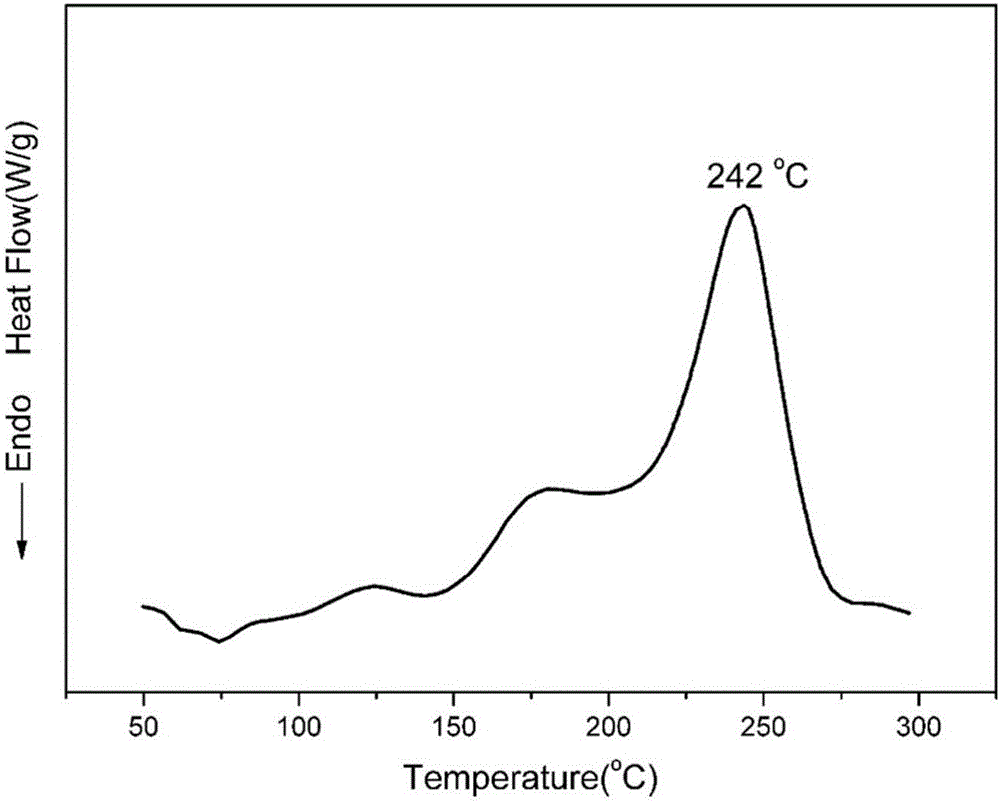
图2实施例1得到的苯并噁嗪齐聚物的DSC图;
图3实施例1得到的苯并噁嗪齐聚物固化后的TGA曲线图
具体实施方式
以下结合具体实例对本发明做进一步描述。有必要指出的是:以下实例只是对本发明做进一步说明,而不仅仅局限于本发明的保护范围。本技术领域的普通技术人员在阅读本发明之后,在不脱离本发明的构思前提下,还可改以做各种改进和调整,这些改进和调整都属于本发明要求保护的范围。
实施例1
(1)将2-氨基苯酚33.4g、马来酸酐30.0g加入到含80mL二甲基甲酰胺(DMF)的冰浴体系中,之后加入向其中加入15g五氧化二磷和8g浓度为95-98%的浓硫酸,反应30分钟后,将反应装置移置油浴中70℃反应6小时。反应结束后,反应液倒入200ml去离子水,得到大量沉淀,反应液经去离子水水洗、过滤、干燥后,得到产物马来酰亚胺官能化酚47.6g,收率82%。反应方程式如下:
(2)称取由上一步所得马来酰亚胺官能化酚30g,以及4,4-二羟基二苯基甲烷(双酚F)47.63g、4,4-二氨基二苯基甲烷62.89g、多聚甲醛38.10g、甲苯150ml分别加入到装有搅拌器,温度计及冷凝管的反应瓶中,以10℃/h的速度加热到120℃反应6h。反应结束后,在反应液中加入去离子水进行沉析,得到大量沉淀,将沉淀物用5%的氢氧化钠溶液洗涤3次,再经水洗,过滤,并在真空下干燥12小时,得到产物129.19g,收率80%。反应方程式如下:
本实施例中,所得苯并噁嗪齐聚物的结构式为:
该产物的傅里叶红外光谱、差示扫描量热法以及固化后树脂的热失重表征结果见附图1、附图2和附图3。附图1为红外光谱图,其中929cm-1处为噁嗪环的特征峰。附图2为差示扫描量热法所得DSC图,可以看出,该齐聚物的最大固化峰值温度为242℃。附图3为固化后树脂的热失重曲线图,可以看出该树脂5%热失重温度为410℃,800℃下的残炭率为70%。
本实施例所得到的苯并噁嗪齐聚物开环固化后,测得其玻璃化转变温度为302℃。
实施例2
(1)将实施例1中的2-氨基苯酚替换为间氨基苯酚,其他步骤同实施例1中的步骤。
其中,间氨基苯酚的结构式为:
(2)在第二步反应中,反应物的量改为:称取由上一步所得马来酰亚胺官能化酚30g,以及4,4-二羟基二苯基甲烷(双酚F)47.63g、4,4-二氨基二苯基甲烷62.89g、多聚甲醛38.10g、甲苯150ml。得到产物135.65g,收率84%。
本实施例中,所得苯并噁嗪齐聚物的结构式为:
本实施例所得到的苯并噁嗪齐聚物开环固化后,测得其玻璃化转变温度为293℃,5%热失重温度为397℃,800℃下的残炭率为68%。
实施例3
(1)将实施例1中的2-氨基苯酚替换为间氨基苯酚,其他步骤同实施例1中的步骤。
其中,对氨基苯酚的结构式为:
(2)在第二步反应中,反应物的量改为:称取马来酰亚胺官能化酚30g,以及4,4-二羟基二苯基甲烷(双酚F)47.63g、4,4-二氨基二苯基甲酮67.32g、多聚甲醛38.10g、二甲苯150ml。得到产物147.67g,收率89%。
本实施例中,所得苯并噁嗪齐聚物的结构式为:
本实施例所得到的苯并噁嗪齐聚物开环固化后,测得其玻璃化转变温度为311℃,5%热失重温度为412℃,800℃下的残炭率为71%。
- 还没有人留言评论。精彩留言会获得点赞!
- 3-(6-溴-4-氧-4H-色酮)-1-苯基-5-对甲氧基苯基-1,6a-二氢吡咯啉[3,4-c]吡唑-4,6 ...的制作方法
- 含有色酮结构的吡唑类n-苯基马来酰亚胺衍生物及其制备方法与应用
- 含有色酮结构的吡唑类n-对甲苯基马来酰亚胺衍生物及其制备方法与应用
- 一种马来酰亚胺丙酰哌嗪七甲川菁盐荧光载体及其制备方法和应用
- 一种利用固体酸催化剂制备n-苯基马来酰亚胺的方法
- 一种n-(取代苯基)马来酰亚胺接枝埃洛石纳米管的制备方法
- 3-(6-溴-4-氧-4H-色酮)-1-苯基-5-对氯苯基-1,6a-二氢吡咯啉[3,4-c]吡唑-4,6(3aH ...的制作方法
- 一种4-(3-氰苄基)苯基乙酰亚胺的合成方法
- 马来酰亚胺封端的含硫聚合物、其组合物及其用图
- 一种提高n-苯基马来酰亚胺收率的方法