包含全甲基并环戊二烯配体的催化剂的制作方法
本发明涉及催化组合物。更具体地,本发明涉及特定的茂金属催化组合物及这种组合物在烯烃聚合反应中的应用。更加具体地,本发明涉及包含具有全甲基并环戊二烯配体的茂金属化合物的组合物及这种组合物在乙烯聚合反应中的应用。
背景技术:
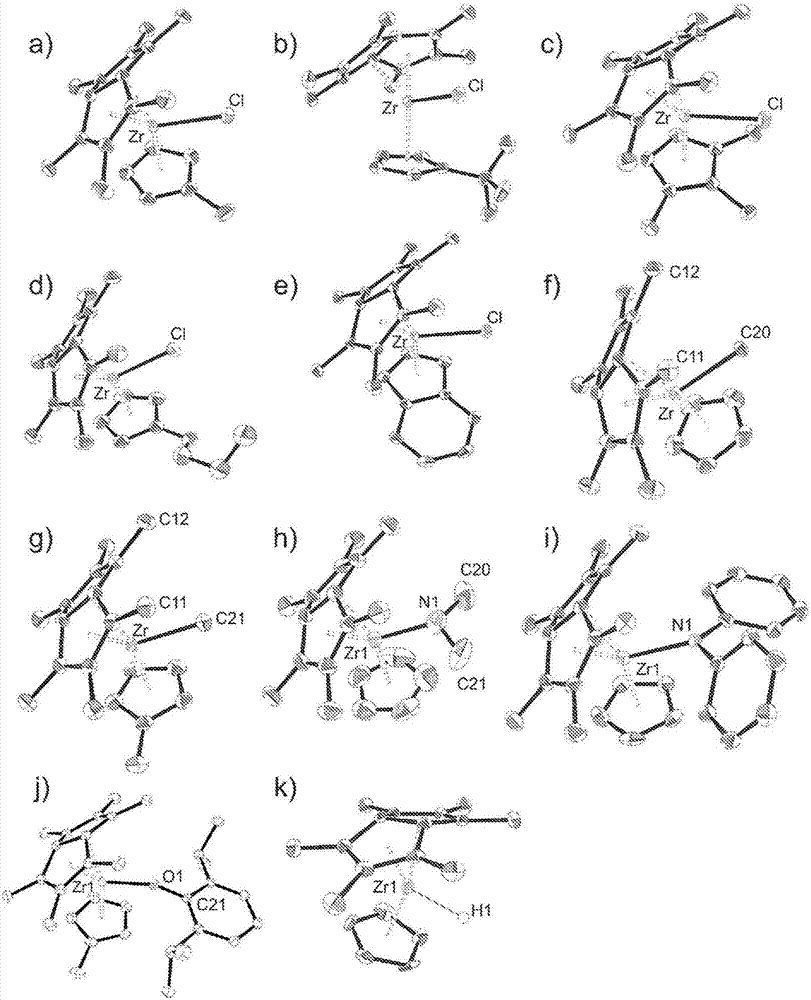
:众所周知在存在某些过渡金属催化剂的情况下在低压或中压下可以容易地聚合乙烯(和一般而言α-烯烃)。这些催化剂通常被称为齐格勒纳塔(Zeigler-Natta)类催化剂。催化乙烯(并且一般为α-烯烃)的聚合的这些齐格勒纳塔类催化剂的特定组包含铝氧烷活化剂和茂金属过渡金属催化剂。茂金属包含两个η5-环戊二烯类配体之间的金属键。许多的茂金属催化剂是本领域已知的。然而,仍需要用于烯烃聚合反应的改善的茂金属催化剂。特别是仍需要具有高聚合活性/效率的新型茂金属催化剂。本发明着眼于以上设计。技术实现要素:根据本发明的第一方面,提供了包含固体甲基铝氧烷载体材料和本文定义的式I的化合物的组合物。根据本发明的进一步的方面,提供了本文定义的组合物在烯烃聚合中的应用。根据本发明的进一步方面,提供了用于聚合一种或多种烯烃的方法,所述方法包括在以下各项存在的情况下聚合一种或多种烯烃的步骤:(i)本文定义的组合物;和(ii)合适的活化剂。根据本发明的另一个方面,提供了通过本文定义的方法可得到、得到或直接得到的聚合物。具体实施方式定义如在本文中使用的术语“烷基”包括涉及通常具有1、2、3、4、5或6个碳原子的直链或支链烷基部分。该术语包括涉及基团诸如甲基、乙基、丙基(正丙基或异丙基)、丁基(正丁基、仲丁基或叔丁基)、戊基(包括新戊基)、己基等。特别地,烷基可以具有1、2、3或4个碳原子。如在本文中使用的术语“烯基”包括涉及通常具有2、3、4、5或6个碳原子的直链或支链烯基部分。术语包括涉及包含1、2或3个碳-碳双键(C=C)的烯基部分。该术语包括涉及基团诸如乙烯基(ethenyl,vinyl)、丙烯基(烯丙基)、丁烯基、戊烯基和己烯基以及它们的顺反异构体两者。如在本文中使用的术语“炔基”包括涉及通常具有2、3、4、5或6个碳原子的直链或支链炔基部分。术语包括涉及包含1、2或3个碳-碳三键(C≡C)的炔基部分。该术语包括涉及基团诸如乙炔基、丙炔基、丁炔基、戊炔基和己炔基。如在本文中使用的术语“烷氧基”包括涉及-O-烷基,其中,烷基是直链或支链并包含1、2、3、4、5或6个碳原子。在一类实施方式中,烷氧基具有1、2、3或4个碳原子。该术语包括涉及基团诸如甲氧基、乙氧基、丙氧基、异丙氧基、丁氧基、叔丁氧基、戊氧基、己氧基等。如在本文中使用的术语“芳基”包括涉及包含6、7、8、9或10个环碳原子的芳环体系。芳基经常是苯基但是可以是多环体系,具有两个或更多个环,其中的至少一个是芳香族的。该术语包括涉及基团诸如苯基、萘基等。如在本文中使用的术语“碳环基”包括涉及具有3、4、5、6、7或8个碳原子的脂环族部分。基团可以是桥接或多环体系。环烷基基团更经常是单环。该术语包括涉及基团诸如环丙基、环丁基、环戊基、环己基、降冰片基、双环[2.2.2]辛基等。如在本文中使用的术语“杂环基”包括涉及具有3、4、5、6、7、8、9或10个环原子的饱和的(例如杂环烷基)或不饱和的(例如杂芳基)杂环部分,其中的至少一个选自氮、氧、磷、硅和硫。特别地,杂环基包括3元至10元环或环体系、以及更特别地5元或6元环,其可以是饱和的或不饱和的。杂环部分例如选自环氧乙烷基、氮丙环基、1,2-氧硫杂环戊基、咪唑基、噻嗯基、呋喃基、四氢呋喃基、吡喃基、硫代吡喃基、噻蒽基、异苯并呋喃基、苯并呋喃基、色烯基、2H-吡咯基、吡咯基、吡咯啉基、吡咯烷基、咪唑浣基、苯并咪唑基、吡唑基、吡嗪基、吡唑烷基、噻唑基、异噻唑基、二噻唑基、噁唑基、异噁唑基、吡啶基、吡嗪基、嘧啶基、哌啶基、哌嗪基、哒嗪基、吗啉基、硫代吗啉基,尤其是硫代吗啉基、吲哚嗪基、异吲哚基、3H-吲哚基、吲哚基、苯并咪唑基、香豆素基(cumaryl)、吲唑基、三唑基、四唑基、嘌呤基、4H-喹嗪基、异喹啉基、喹啉基、四氢喹啉基、四氢异喹啉基、十氢喹啉基、八氢异喹啉基、苯并呋喃基、二苯并呋喃基、苯并苯硫基、二苯并苯硫基、酞嗪基、萘啶基、喹喔啉基、喹唑啉基、喹唑啉基、噌啉基、蝶啶基、咔唑基、β-咔啉基、菲啶基、吖啶基、萘嵌间二氮苯基、磷二氮杂菲基、呋咱基、吩嗪基、吩噻嗪基、吩噁嗪基、色烯基、异色满基、色满基等。如在本文中使用的术语“杂芳基”包括涉及具有5、6、7、8、9或10个环原子的芳香族杂环体系,其中的至少一个选自氮、氧和硫。基团可以是多环体系,具有两个或更多个环,其中的至少一个是芳香族,但是更经常是单环的。该术语包括涉及基团诸如嘧啶基、呋喃基、苯并[b]苯硫基、苯硫基、吡咯基、咪唑基、吡咯烷基、吡啶基、苯并[b]呋喃基、吡嗪基、嘌呤基、氮茚基、苯并咪唑基、喹啉基、吩噻嗪基、三嗪基、酞嗪基、2H-苯并吡喃基(chromenyl)、噁唑基、异噁唑基、噻唑基、异吲哚基、吲唑基、嘌呤基、异喹啉基、间二氮杂萘基、蝶啶基等的基团。如在本文中使用的术语“卤素(halogen)”或“卤(halo)”包括涉及F、Cl、Br或I。特别地,卤素可以是F或Cl,其中Cl是更常见的。如在本文中使用的关于部分的术语“取代的”是指所述部分中的一个或多个、尤其最高达5个、更尤其1、2或3个氢原子彼此独立地被相应数目的所描述的取代基取代。如在本文中使用的术语“可选取代的”是指取代的或未取代的。当然将理解取代基仅在它们化学可能的位置,本领域技术人员能够在适当努力下判定(实践或理论上)特定的取代是否可能。例如,具有游离氢的氨基或羟基基团如果结合至具有不饱和(例如烯属)键的碳原子可能是不稳定的。另外,当然应理解如技术人员公认的,本文所描述的取代基自身可以被任何取代基取代,受上述适当取代的限制。本发明的组合物如在上文中讨论的,本发明提供了包含固体甲基铝氧烷支撑材料和以下所示的式I的化合物的组合物:其中,R1和R2各自独立地是氢或直链(1-4C)烷基,或R1和R2连接使得当与它们所附接的原子结合时,它们形成可选地被选自(1-6C)烷基、(2-6C)烯基、(2-6C)炔基、(1-6C)烷氧基、芳基、杂芳基、碳环和杂环中的一个或多个基团取代的6元稠合芳环,其中每个芳基、杂芳基、碳环和杂环基团可选地被选自(1-6C)烷基、(2-6C)烯基、(2-6C)炔基、(1-6C)烷氧基、卤素、氨基、硝基、氰基、(1-6C)烷氨基、[(1-6C)烷基]2氨基和-S(O)2(1-6C)烷基中的一个或多个基团取代;R3和R4各自独立地是氢或直链(1-4C)烷基,或R3和R4连接使得当与它们所附接的原子结合时,它们形成可选地被选自(1-6C)烷基、(2-6C)烯基、(2-6C)炔基、(1-6C)烷氧基、芳基、杂芳基、碳环和杂环中的一个或多个基团取代的6元稠合芳环,其中每个芳基、杂芳基、碳环和杂环基团可选地被选自(1-6C)烷基、(2-6C)烯基、(2-6C)炔基、(1-6C)烷氧基、卤素、氨基、硝基、氰基、(1-6C)烷氨基、[(1-6C)烷基]2氨基和-S(O)2(1-6C)烷基中的一个或多个基团取代;R5是氢或直链(1-4C)烷基;X选自锆或铪;以及Y选自卤素阴离子(haloanion)、氢阴离子(氢负离子、氢离子,hydrideanion)、膦酸根阴离子(phosphonatedanion)、磺酸根阴离子(sulfonatedanion)或硼酸根阴离子(borateanion)、或(1-6C)烷基、(2-6C)烯基、(2-6C)炔基、(1-6C)烷氧基、-C(O)NRaRb、-NRaRb、(可选地被选自卤素、(1-4C)烷基、硝基、-NRaRb、苯基、(1-6C)烷氧基、-C(O)NRaRb或Si[(1-4C)烷基]3中的一个或多个基团取代的)芳基或芳氧基基团。其中,Ra和Rb独立地是氢或(1-4C)烷基。当与目前的用于α-烯烃的聚合的全甲基并环戊二烯茂金属化合物/组合物比较时,本发明的组合物表现出优异的催化性能。特别地,当与类似的二氧化硅支撑的甲基铝氧烷(SSMAO)催化剂组合物比较时,本发明的固体MAO组合物在α-烯烃的聚合中表现出异乎寻常高的催化活性。固体甲基铝氧烷(MAO)(经常称为聚甲基铝氧烷)与甲基铝氧烷(MAO)的区别在于其不可溶于烃溶剂,所以可以充当合适的烃溶剂中的异质支撑体系。可以使用任何合适的固体MAO载体。术语“固体甲基铝氧烷”、“固体MAO”和“固体聚甲基铝氧烷”在本文中同义使用,是指具有通式-[(Me)AlO]n-的固相材料,其中,n是4至50的整数(例如10至50)。可以使用任何合适的固体聚甲基铝氧烷。固体聚甲基铝氧烷和其他(非固体)MAO之间存在许多显著的结构上和行为上的差异。或许最值得注意的是,固体聚甲基铝氧烷与其他MAO的区别在于其不可溶于烃溶剂,从而充当异质载体体系。可用于本发明的组合物的固体聚甲基铝氧烷不可溶于甲苯和己烷。与非固体(可溶于烃)MAO(其传统用作浆液聚合中的活化剂物质或用于改性单独的固体载体材料(例如SiO2)的表面)不同,可用作本发明的一部分的固体聚甲基铝氧烷本身适合用作固相载体材料,不需要另外的活化剂。因此,包含固体聚甲基铝氧烷的本发明的组合物不含可以被称为固体载体(例如无机材料诸如SiO2、Al2O3和ZrO2)的任何其他物质。此外,考虑到固体聚甲基铝氧烷的双重功能(作为催化载体和活化剂物质),包含固体MAO的本发明的组合物可以不包含另外的催化活化剂物质。在一个实施方式中,通过加热包含MAO和烃溶剂(例如甲苯)的溶液制备固体聚甲基铝氧烷,从而使固体聚甲基铝氧烷沉淀。可以通过在烃溶剂(例如甲苯)中使三甲基铝和苯甲酸反应,然后加热生成的混合物制备包含MAO和烃溶剂的溶液。在一个实施方式中,根据以下流程制备固体聚甲基铝氧烷。可以通过改变其合成期间使用的加工变量中的一个或多个调节固体聚甲基铝氧烷的性质。例如,在以上列出的流程中,可以通过改变Al:O比值,通过固定AlMe3的量和改变苯甲酸的量调节固体聚甲基铝氧烷的性质。示例性的Al:O比值是1:1、1.1:1、1.2:1、1.3:1、1.4:1和1.6:1。适当地,Al:O比值是1.2:1或1.3:1。可替换地,可以通过固定苯甲酸的量和改变AlMe3的量来调节固体聚甲基铝氧烷的性质。在一个实施方式中,根据以下流程制备固体聚甲基铝氧烷。在以上流程中,可以保持步骤1和2不变,改变步骤2。步骤2的温度可以是70-100℃(例如70℃、80℃、90℃或100℃)。步骤2的持续时间可以是12至28小时(例如12、20或28小时)。步骤2的持续时间可以是5分钟至24小时。可以在溶剂诸如甲苯中进行步骤3。在一个实施方式中,固体聚甲基铝氧烷的铝含量在36-41wt%的范围内。可用作本发明的一部分的固体聚甲基铝氧烷的特征在于在甲苯和正己烷中非常低的溶解度。在一个实施方式中,固体聚甲基铝氧烷在25℃的正己烷中的溶解度是0-2mol%。适当地,固体聚甲基铝氧烷在25℃的正己烷中的溶解度是0-1mol%。更适当地,固体聚甲基铝氧烷在25℃的正己烷中的溶解度是0-0.2mol%。可替换地或另外地,固体聚甲基铝氧烷在25℃的甲苯中的溶解度是0-2mol%。适当地,固体聚甲基铝氧烷在25℃的甲苯中的溶解度是0-1mol%。更适当地,固体聚甲基铝氧烷在25℃的甲苯中的溶解度是0-0.5mol%。可以通过JP-B(KOKOKU)-H0742301中描述的方法测量在溶剂中的溶解度。在一个特别合适的实施方式中,US2013/0059990、WO2010/055652或WO2013/146337中描述了固体聚甲基铝氧烷,并可由日本的TosohFinechemCorporation得到。在一个实施方式中,固体聚甲基铝氧烷与式(I)的化合物的摩尔比是50:1至500:1。适当地,固体聚甲基铝氧烷与式(I)的化合物的摩尔比是75:1至400:1。更适当地,固体聚甲基铝氧烷与式(I)的化合物的摩尔比是100:1至300:1。在一个实施方式中,固体MAO载体不可溶于甲苯和己烷。在另一个实施方式中,固体MAO载体是以颗粒形式。适当地,固体MAO载体的颗粒的形状是球形的、或基本上球形的。可以通过一种或多种离子或共价相互作用将式(I)的化合物固定在固体MAO载体上。在一个实施方式中,组合物进一步包含一种或多种合适的活化剂。合适的活化剂是本领域众所周知的并包括有机铝化合物(例如烷基铝化合物)。特别合适的活化剂包括铝氧烷(例如甲基铝氧烷(MAO))、三异丁基铝(TIBA)、二乙基铝(DEAC)和三乙基铝(TEA)。一种或多种活化剂可以采取能够摄取氧、水和其他质子杂质中的一种或多种的物质的形式。在另一个实施方式中,固体MAO载体包含选自M(C6F5)3(其中M是铝或硼)或M’R2(其中M’是锆或镁以及R是(1-10C)烷基(例如甲基或辛基))中的至少一种另外的化合物。在一个实施方式中,当Y是Cl时,R1、R2、R3、R4和R5中的至少一个是H之外的基团。在一个实施方式中,R1、R2、R3、R4和R5中的至少一个是H之外的基团。在另一个实施方式中,组合物不包含具有以下结构中的任一种的式I的化合物:在另一个实施方式中,R1和R2各自独立地是氢或直链(1-4C)烷基,或R1和R2连接使得当与它们所附接的原子结合时,它们形成可选地被选自(1-6C)烷基、(2-6C)烯基、(2-6C)炔基、(1-6C)烷氧基、芳基、杂芳基、碳环和杂环中的一个或多个基团取代的6元稠合芳环,其中,每个芳基、杂芳基、碳环和杂环基团可选地被选自(1-6C)烷基、(2-6C)烯基、(2-6C)炔基、(1-6C)烷氧基和卤素中的一个或多个基团取代。适当地,R1和R2各自独立地是氢或直链(1-4C)烷基,或R1和R2连接使得当与它们所附接的原子结合时,形成可选地被选自(1-6C)烷基、(2-6C)烯基、(2-6C)炔基、(1-6C)烷氧基、芳基或杂芳基中的一个或多个基团取代的6元稠合芳环,其中,每个芳基或杂芳基基团可选地被选自(1-6C)烷基、(2-6C)烯基、(2-6C)炔基、(1-6C)烷氧基和卤素中的一个或多个基团取代。更适当地,R1和R2各自独立地是氢或直链(1-4C)烷基,或R1和R2连接使得当与它们所附接的原子结合时,它们形成可选地被选自(1-4C)烷基、(2-4C)烯基、(2-4C)炔基、(1-4C)烷氧基、芳基或杂芳基中的一个或多个基团取代的6元稠合芳环,其中,每个芳基或杂芳基基团可选地被选自(1-4C)烷基、(2-4C)烯基、(2-4C)炔基、(1-4C)烷氧基和卤素中的一个或多个基团取代。甚至更适当地,R1和R2各自独立地是氢或直链(1-4C)烷基,或R1和R2连接使得当与它们所附接的原子结合时,它们形成6元稠合芳环。甚至更适当地,R1和R2各自独立地是氢、甲基或正丁基,或R1和R2连接使得当与它们所附接的原子结合时,它们形成6元稠合芳环。在另一个实施方式中,R3和R4各自独立地是氢或直链(1-4C)烷基,或R1和R2连接使得当与它们所附接的原子结合时,它们形成可选地被选自(1-6C)烷基、(2-6C)烯基、(2-6C)炔基、(1-6C)烷氧基、芳基、杂芳基、碳环和杂环中的一个或多个基团取代的6元稠合芳环,其中,每个芳基、杂芳基、碳环和杂环基团可选地被选自(1-6C)烷基、(2-6C)烯基、(2-6C)炔基、(1-6C)烷氧基和卤素中的一个或多个基团取代。适当地,R3和R4各自独立地是氢或直链(1-4C)烷基,或R1和R2连接使得当与它们所附接的原子结合时,它们形成可选地被选自(1-6C)烷基、(2-6C)烯基、(2-6C)炔基、(1-6C)烷氧基、芳基或杂芳基中的一个或多个基团取代的6元稠合芳环,其中,每个芳基或杂芳基基团可选地被选自(1-6C)烷基、(2-6C)烯基、(2-6C)炔基、(1-6C)烷氧基和卤素中的一个或多个基团取代。更适当地,R3和R4各自独立地是氢或直链(1-4C)烷基,或R1和R2连接使得当与它们所附接的原子结合时,它们形成可选地被选自(1-4C)烷基、(2-4C)烯基、(2-4C)炔基、(1-4C)烷氧基、芳基或杂芳基中的一个或多个基团取代的6元稠合芳环,其中,每个芳基或杂芳基基团可选地被选自(1-4C)烷基、(2-4C)烯基、(2-4C)炔基、(1-4C)烷氧基和卤素中的一个或多个基团取代。甚至更适当地,R3和R4各自独立地是氢或直链(1-4C)烷基,或R1和R2连接使得当与它们所附接的原子结合时,它们形成6元稠合芳环。甚至更适当地,R3和R4各自独立地是氢、甲基或正丁基,或R3和R4连接使得当与它们所附接的原子结合时,它们形成6元稠合芳环。在另一个实施方式中,R5是氢、甲基或正丁基。在另一个实施方式中,R5是氢或甲基。适当地,R5是氢。在另一个实施方式中,X是锆。Y选自卤素阴离子(haloanion)、氢阴离子(氢负离子,hydrideanion)、酰胺阴离子(amideanion)、膦酸根阴离子(phosphonatedanion)、磺酸盐根阴离子(sulfonatedanion)或硼酸根阴离子(borateanion)、或(1-6C)烷基、(2-6C)烯基、(2-6C)炔基、(1-6C)烷氧基、-C(O)NRaRb、-NRaRb、(可选地被选自卤素、(1-4C)烷基、硝基、-NRaRb、苯基、(1-6C)烷氧基、-C(O)NRaRb或Si[(1-4C)烷基]3中的一个或多个基团取代的)芳基或芳氧基基团。适当地,Y是-NRaRb,其中Ra和Rb都是氢以及都被苯基取代得到基团-N(C6H5)2。在另一个实施方式中,Y选自卤素阴离子、氢阴离子、酰胺阴离子、膦酸根阴离子、磺酸盐根阴离子硼酸根阴离子、或(1-6C)烷基、(2-6C)烯基、(2-6C)炔基、(1-6C)烷氧基、-NRaRb、(可选地被选自卤素、(1-4C)烷基和苯基中的一个或多个基团取代的)芳基或芳氧基基团。在另一个实施方式中,Y选自卤素阴离子、氢阴离子、膦酸根阴离子、磺酸根阴离子或硼酸根阴离子、或(1-6C)烷基、(2-6C)烯基、(2-6C)炔基、(1-6C)烷氧基、-NRaRb、可选地被选自卤素、(1-4C)烷基和苯基中的一个或多个基团取代的芳基或芳氧基基团。适当地,Y选自卤素、氢阴离子(氢化物,hydride)、或(1-6C)烷基、(2-6C)烯基、(2-6C)炔基、(1-6C)烷氧基、-NRaRb、(可选地被选自卤素、(1-4C)烷基和苯基中的一个或多个基团取代的)芳基或芳氧基基团。更适当地,Y选自卤素、氢阴离子、或(1-4C)烷基、(1-5C)烷氧基、-NRaRb、(可选地被选自卤素、(1-4C)烷基和苯基中的一个或多个基团取代的)芳基或芳氧基基团。甚至更适当地,Y是卤素、氢阴离子、甲基、正丁基、-N(CH3)2、-N(C6H5)2、-O-2,6-二甲基-C6H3)、-O-2,6-二异丙基-C6H3)、-O-2,4-二叔丁基-C6H3)、-O-C(CH3)2CH2CH3。仍更适当地,Y是Cl或甲基。最适当地,Y是甲基。在另一个实施方式中,Y选自卤素阴离子、氢阴离子、膦酸根阴离子、磺酸根阴离子或硼酸根阴离子、或(1-6C)烷基、(2-6C)烯基、(2-6C)炔基、(1-6C)烷氧基、(可选地被选自卤素和(1-4C)烷基中的一个或多个基团取代的)芳基或芳氧基基团。适当地,Y选自卤素、氢阴离子、或(1-6C)烷基、(2-6C)烯基、(2-6C)炔基、(1-6C)烷氧基、(可选地被选自卤素和(1-4C)烷基中的一个或多个基团取代的)芳基或芳氧基基团。更适当地,Y选自卤素、氢阴离子、或(1-4C)烷基、(2-4C)烯基、(2-4C)炔基、(1-4C)烷氧基、(可选地被选自卤素和(1-4C)烷基中的一个或多个基团取代的)芳基或芳氧基基团。甚至更适当地,Y是卤素。仍更适当地,Y是Cl、Br或I。最适当地,Y是Cl。在另一个实施方式中,式I的化合物具有根据以下所示的式Ia、Ib,或Ic的结构:其中,R1和R2各自独立地是氢或直链(1-4C)烷基,R3和R4各自独立地是氢或直链(1-4C)烷基,Rx各自独立地选自(1-6C)烷基、(2-6C)烯基、(2-6C)炔基、(1-6C)烷氧基、芳基、杂芳基、碳环和杂环,其中每个芳基、杂芳基、碳环和杂环基团可选地被选自(1-6C)烷基、(2-6C)烯基、(2-6C)炔基、(1-6C)烷氧基、卤素、氨基、硝基、氰基、(1-6C)烷氨基、[(1-6C)烷基]2氨基和-S(O)2(1-6C)烷基中的一个或多个基团取代;n各自独立地是选自0、1、2、3,或4的整数;X是Zr或Hf;以及Y选自卤素阴离子、氢阴离子、膦酸根阴离子、磺酸根阴离子或硼酸根阴离子、或(1-6C)烷基、(2-6C)烯基、(2-6C)炔基、(1-6C)烷氧基、-C(O)NRaRb、-NRaRb、(可选地被选自卤素、(1-4C)烷基、硝基、-NRaRb、苯基、(1-6C)烷氧基、-C(O)NRaRb或Si[(1-4C)烷基]3中的一个或多个基团取代的)芳基或芳氧基基团。其中,Ra和Rb独立地是氢或(1-4C)烷基。在另一个实施方式中,式I的化合物具有根据式Ia、Ib,或Ic的结构,其中,Rx各自独立地选自(1-6C)烷基、(2-6C)烯基、(2-6C)炔基和(1-6C)烷氧基;以及n各自独立地是选自0、1或2的整数。在另一个实施方式中,式I的化合物具有根据式Ia、Ib或Ic的结构,其中,X是Zr;以及Y选自卤素、氢阴离子、或(1-6C)烷基、(2-6C)烯基、(2-6C)炔基、(1-6C)烷氧基、-NRaRb、可选地被选自卤素、(1-4C)烷基和苯基中的一个或多个基团取代的芳基或芳氧基基团;其中,Ra和Rb独立地是氢或甲基。在另一个实施方式中,式I的化合物具有根据式Ia、Ib或Ic的结构,其中,R1和R2各自独立地是氢或直链(1-2C)烷基;R3和R4各自独立地是氢或直链(1-2C)烷基;Rx各自独立地选自(1-3C)烷基、(2-3C)烯基、(2-3C)炔基和(1-3C)烷氧基;n各自独立地是选自0、1或2的整数;X是Zr;以及Y选自卤素、氢阴离子、或(1-6C)烷基、(1-5C)烷氧基或(可选地被选自卤素或(1-4C)烷基中的一个或多个基团取代的)芳氧基基团,或Y是基团-N(CH3)2或-N(C6H5)2。在另一个实施方式中,式I的化合物具有根据式Ia、Ib或Ic的结构,其中,R1和R2各自独立地是氢或直链(1-4C)烷基;R3和R4各自独立地是氢或直链(1-4C)烷基;n是0;X是Zr或Hf;以及Y选自卤素、氢阴离子、或(1-6C)烷基、(1-5C)烷氧基或(可选地被选自卤素或(1-4C)烷基中的一个或多个基团取代的)芳氧基基团,或Y是基团-N(CH3)2或-N(C6H5)2。在另一个实施方式中,式I的化合物具有根据式Ia、Ib或Ic的结构,其中,R1和R2各自独立地是氢或(1-2C)烷基;R3和R4各自独立地是氢或(1-2C)烷基;n是0;X是Zr或Hf;以及Y选自卤素、氢阴离子、或(1-6C)烷基或(可选地被选自卤素或(1-4C)烷基中的一个或多个基团取代的)芳氧基基团,或Y是基团-N(CH3)2或-N(C6H5)2。在另一个实施方式中,式I的化合物具有根据式Ia、Ib或Ic的结构,其中,R1和R2各自独立地是氢、甲基或正丁基;R3和R4各自独立地是氢、甲基或正丁基;n是0;X是Zr或Hf;以及Y选自卤素、氢阴离子、或(1-6C)烷基(例如甲基或正丁基)或(可选地被选自卤素或(1-4C)烷基中的一个或多个基团取代的)芳氧基基团,或Y是基团-N(CH3)2或-N(C6H5)2。在另一个实施方式中,式I的化合物具有根据式Ia、Ib或Ic的结构,其中,Rx各自独立地选自(1-6C)烷基、(2-6C)烯基、(2-6C)炔基和(1-6C)烷氧基;以及n各自独立地是选自0、1或2的整数。在另一个实施方式中,式I的化合物具有根据式Ia、Ib或Ic的结构,其中,X是Zr;以及Y选自卤素、氢阴离子、或(1-6C)烷基、(2-6C)烯基、(2-6C)炔基、(1-6C)烷氧基、(可选地被选自卤素或(1-4C)烷基中的一个或多个基团取代的)芳基或芳氧基基团。在另一个实施方式中,式I的化合物具有根据式Ia、Ib或Ic的结构,其中,R1和R2各自独立地是氢或直链(1-2C)烷基;R3和R4各自独立地是氢或直链(1-2C)烷基;Rx各自独立地选自(1-3C)烷基、(2-3C)烯基、(2-3C)炔基和(1-3C)烷氧基;n各自独立地是选自0、1或2的整数;X是Zr;以及Y选自卤素、氢阴离子、或(1-6C)烷基或(可选地被选自卤素或(1-4C)烷基中的一个或多个基团取代的)芳氧基基团。在另一个实施方式中,式I的化合物具有根据式Ia、Ib或Ic的结构,其中,R1和R2各自独立地是氢或直链(1-4C)烷基;R3和R4各自独立地是氢或直链(1-4C)烷基;n是0;X是Zr或Hf;以及Y选自卤素、氢阴离子、或(1-6C)烷基或(可选地被选自卤素或(1-4C)烷基中的一个或多个基团取代的)芳氧基基团。在另一个实施方式中,式I的化合物具有根据式Ia、Ib或Ic的结构,其中,R1和R2各自独立地是氢或(1-2C)烷基;R3和R4各自独立地是氢或(1-2C)烷基;n是0,X是Zr或Hf;以及Y选自卤素、氢阴离子、或(1-6C)烷基或(可选地被选自卤素或(1-4C)烷基中的一个或多个基团取代的)芳氧基基团。在另一个实施方式中,式I的化合物具有根据式Ia、Ib或Ic的结构,其中,R1和R2各自独立地是氢或甲基;R3和R4各自独立地是氢或甲基;n是0;X是Zr或Hf;以及Y选自卤素、氢阴离子、或(1-6C)烷基或(可选地被选自卤素或(1-4C)烷基中的一个或多个基团取代的)芳氧基基团。在一个实施方式中,式I的化合物具有以下结构中的任一种:在一个具体的实施方式中,式I的化合物具有以下结构中的任一种:在另一个具体的实施方式中,式I的化合物具有以下结构中的任一种:在另一个具体的实施方式中,式I的化合物具有以下结构中的任一种:在一个具体的实施方式中,式I的化合物具有以下结构中的任一种:适当地,式I的化合物具有以下结构:可替换地,式I的化合物具有以下结构中的任一种:合成通过本领域已知的任何合适的方法合成可用作本发明的一部分的化合物。在所附实施例中阐述了用于制备上述化合物的方法的具体实例。适当地,根据以下方案1制备化合物。方案1:合成Pn*ZrCpRCl的一般合成途径的一个实例关于以上方案1,将理解五个“R”基团分别具有根据本文定义的R1、R2、R3、R4和R5的定义。将了解“Cl”仅是本文定义的Y基团的一个实例,而且技术人员将容易了解如何将Cl更换为本文定义的其他Y基团。类似地,将了解可以将“Li”更换为替代金属。可以使用任何合适的溶剂。在必要时,本领域技术人员将能够选择用于这种合成的合适的反应条件(例如温度、压力、反应时间、搅拌等)。在以下方案2-8中阐述了本发明涵盖的得到其他前体催化剂的示例性合成途径。方案2:合成Pn*ZrCpR(Me)的一般合成途径方案3:合成Pn*ZrCpRH的一般合成途径方案4:合成Pn*ZrCpR(nBu)的一般合成途径方案5:合成Pn*ZrCpR(NR’2)的一般合成途径方案6:合成Pn*ZrCpR(O-2,6-R’-C6H3)的一般合成途径方案7:合成Pn*ZrCpMe(O-2,4-tBu-C6H3)的合成途径方案8:合成Pn*ZrCpMe(OCMe2Et)的合成途径制备后,可以通过在合适的溶剂(例如甲苯)中在可选的加热情况下使式(I)的化合物与本文定义的固体MAO接触,然后分离产生的固体将所述式的化合物转化为本发明的组合物。在所附实施例中阐述了用于制备本发明的式(I)的化合物的示例性过程。本文还描述了本文定义的、通过本文定义的方法可得到的、得到的或直接得到的组合物。应用如上文所讨论的,本发明的组合物是烯烃聚合中有效的催化剂/引发剂。因此,如上文中讨论的,本发明还提供了本文定义的组合物在烯烃聚合中的应用。在一个实施方式中,烯烃都是乙烯(ethene,ethylene),因此产生聚乙烯均聚物。在另一个实施方式中,烯烃是不同的,从而产生共聚物。在一个实施方式中,烯烃的混合物包含90-99wt%的乙烯单体和1-10wt%的(4-8C)α-烯烃。适当地,(4-8C)α-烯烃是1-丁烯、1-己烯、1-辛烯或它们的混合物。如上文所讨论的,本发明还提供了用于聚合一种或多种烯烃的方法,所述方法包括在以下各项存在的情况下聚合一种或多种烯烃的步骤:(i)本文定义的组合物;和(ii)合适的活化剂。合适的活化剂是本领域众所周知的并包括有机铝化合物(例如烷基铝化合物)。特别合适的活化剂包括铝氧烷(例如甲基铝氧烷(MAO))、三异丁基铝(TIBA)、二乙基铝(DEAC)和三乙基铝(TEA)。在另一个实施方式中,烯烃是乙烯单体,从而产生聚乙烯均聚产物。在另一个实施方式中,烯烃是烯烃的混合物,从而产生共聚产物。烯烃的混合物可以包含90-99wt%的乙烯单体和1-10wt%的(4-8C)α-烯烃。适当地,(4-8C)α-烯烃是1-丁烯、1-己烯、1-辛烯或它们的混合物。烯烃聚合领域的技术人员将能够选择用于这种聚合反应的合适的反应条件(例如温度、压力、反应时间等)。本领域技术人员将能够操控过程参数以生产具有特定性质的聚烯烃。在一个实施方式中,在30-90℃的温度下进行所述方法。适当地,在35-65℃的温度下进行所述方法。形成所述方法的一部分的合适的活化剂相对于本发明的组合物可以具有出现在上文中的那些定义中的任一种。将了解形成所述方法的一部分的合适的活化剂可以固有的存在于组合物自身内,使得在仅存在1种类型的活化剂的情况下进行所述方法。可替换地,除了固有地存在于组合物中的活化剂之外,可以存在形成所述方法的一部分的合适的活化剂,使得事实上可以在存在2种类型的活化剂的情况下进行所述方法。任一活化剂可以是摄氧物质的形式。实施例仅出于示出的目的,将参照附图描述本发明的实施例,其中:图1示出了A)通过X射线晶体分析法确定的(左)Pn*ZrCp1,2,3-MeCl;(右)Pn*ZrIndCl的分子结构。在50%概率示出了热椭圆体。B)通过X射线晶体分析法确定的a)Pn*ZrCpMeCl、b)Pn*ZrCptBuCl、c)Pn*ZrCpMe3Cl、d)Pn*ZrCpnBuCl、e)Pn*ZrIndCl、f)Pn*ZrCpMe、g)Pn*ZrCpMe(Me)、h)Pn*ZrCp(NMe2)、i)Pn*ZrCp(NPh2)、j)Pn*ZrCpMe(O-2,6-Me-C6H3)和k)Pn*ZrCp(H)的分子结构。图2示出了各种固体MAO支撑的Pn*ZrCpRCl复合物(络合物,complex)在乙烯的浆液相聚合中的活性。聚合条件:1:200的[Zr]:[TIBA],50mL甲苯,60℃,2巴和30分钟。图3示出了支撑在聚甲基铝氧烷(固体MAO)上的前体催化剂Pn*ZrCpRCl(CpR=Cp、CpMe、CptBu、CpnBu、CpMe3、Ind)和Pn*ZrCpMeMe的乙烯聚合活性。聚合条件:[Zr]:[sMAO]=1:200;150mgTiBA助催化剂;2巴乙烯;10mg催化剂负荷;50mL甲苯;60℃;30分钟。图4示出了在Pn*ZrCpRCl(CpR=Cp、CpMe、CptBu、CpMe3、Ind)的情况下聚甲基铝氧烷支撑的60℃下乙烯聚合的聚合物分子量,Mw。在括号中给出了PDI。聚合条件:[Zr]:[sMAO]=1:200;150mgTiBA助催化剂;2巴乙烯;10mg催化剂负荷;50mL甲苯;60℃;30分钟。图5示出了通过GPC确定的由聚甲基铝氧烷支撑的Pn*ZrCptBuCl产生的聚合物的分子量分布。图6示出了在Pn*ZrCpMeCl的情况下乙烯聚合活性的温度依赖性。聚合条件:[Zr]:[sMAO]=1:200;150mgTiBA助催化剂;2巴乙烯;10mg催化剂负荷;50mL甲苯;30分钟。图7示出了在Pn*ZrCpnBuCl的情况下乙烯聚合活性的温度依赖性。聚合条件:[Zr]:[sMAO]=1:200;150mgTiBA助催化剂;2巴乙烯;10mg催化剂负荷;50mL甲苯;30分钟。图8示出了在Pn*ZrCpMe(Me)的情况下乙烯聚合活性的温度依赖性。聚合条件:[Zr]:[sMAO]=1:200;150mgTiBA助催化剂;2巴乙烯;10mg催化剂负荷;50mL甲苯;30分钟图9示出了在聚甲基铝氧烷支撑的前体催化剂Pn*ZrCpCl的情况下乙烯聚合活性和聚合物分子量MW的时间依赖性。在括号中给出了PDI。聚合条件:[Zr]:[sMAO]=1:200;150mgTiBA助催化剂;2巴乙烯;10mg催化剂负荷;50mL甲苯;60℃。图10示出了在聚甲基铝氧烷支撑的前体催化剂Pn*ZrCpCl的情况下乙烯聚合活性和聚合物分子量MW的温度依赖性。在括号中给出了PDI。聚合条件:[Zr]:[sMAO]=1:200;150mgTiBA助催化剂;2巴乙烯;10mg催化剂负荷;50mL甲苯。图11示出了在Pn*ZrCp(NMe2)的情况下乙烯聚合活性的温度依赖性。聚合条件:[Zr]:[sMAO]=1:200;150mgTiBA助催化剂;2巴乙烯;10mg催化剂负荷;50mL甲苯;30分钟。图12示出了在Pn*ZrCp(NPh2)的情况下乙烯聚合活性的温度依赖性。聚合条件:[Zr]:[sMAO]=1:200;150mgTiBA助催化剂;2巴乙烯;10mg催化剂负荷;50mL甲苯;30分钟。图13示出了在聚甲基铝氧烷支撑的前体催化剂Pn*ZrCp(NMe2)的情况下乙烯聚合活性的时间依赖性。聚合条件:[Zr]:[sMAO]=1:200;150mgTiBA助催化剂;2巴乙烯;10mg催化剂负荷;50mL甲苯;40℃。图14示出了在聚甲基铝氧烷支撑的前体催化剂Pn*ZrCp(NPh2)的情况下乙烯聚合活性的时间依赖性。聚合条件:[Zr]:[sMAO]=1:200;150mgTiBA助催化剂;2巴乙烯;10mg催化剂负荷;50mL甲苯;40℃。图15示出了使用c)×100放大率下的Pn*ZrCpMeCl;d)×250放大率下的Pn*ZrCpMeCl;e)×100放大率下的Pn*ZrCptBuCl;f)×250放大率下的Pn*ZrCptBuCl;g)×100放大率下的Pn*ZrIndCl;和h)×250放大率下的Pn*ZrIndCl的乙烯的浆液相聚合产生的聚合物的SEM。浆液相聚合条件:[Zr]:[MAO]=1:200;150mgTiBA助催化剂;2巴乙烯;10mg催化剂负荷;50mL甲苯;30分钟。实施例1-合成前体催化剂实施例1a-合成Pn*ZrCpMeCl将ZrPn*(μ-Cl)3/2]2(μ-Cl)2Li.Et2O(1.21)(300mg,0.362mmol)溶解在Et2O(20mL)中并冷却至-78℃。将Et2O(15mL)添加到LiCpMe(62.3mg,0.724mmol)中,冷却至-78℃,并将内容物浆液添加到[ZrPn*(μ-Cl)3/2]2(μ-Cl)2Li.Et2O(1.21)的溶液上。允许在一小时内将Schlenk升至室温,然后搅拌另一小时。然后在真空下除去Et2O,以及在过滤到小的schlenk之前将固体少量溶解在苯(3×2mL)中。将苯冷冻在-78℃,从冷水浴除去,并暴露于动态真空过夜,导致溶剂升华。然后用-78℃戊烷(2×3mL)洗涤固体。以及在真空下干燥4h,给出54%产率的产物(152mg,0.388mmol)。1HNMR(C6D6)δ(ppm):1.66{(2,6)-Me2,6H,s};1.81{(3,5)-Me2,6H,s};2.12{(1,7)-Me2,Cp-Me,9H,s);5.13{Cp(2,5)-CH,2H,t(3JH-H=2.6Hz)};5.59{Cp(3,4)-CH,2H,t(3JH-H=2.6Hz)}13CNMR(C6D6)δ(ppm):10.8{(2,6)-Me2};12.4{(1,7)-Me2};13.0{(3,5)-Me2};14.5(Cp-Me);106.1{Cp(2,5)};113.6{Cp(3,4)}124.3(Cp(1)}实施例1b-合成ZrPn*CpMe3Cl将[ZrPn*(μ-Cl)3/2]2(μ-Cl)2Li.THF(1.02)(250mg,0.308mmol)溶解在Et2O(20mL)中并冷却至-78℃。经由套管将1,2,3-CpMe3(70.2mg,0.615mmol)在-78℃Et2O(15mL)中的浆液转移到该溶液中,并在该温度下搅拌内容物1h。然后将Schlenk缓慢升至室温,之后搅拌另一小时。在真空中除去溶剂,将内容物溶解在苯(3×2mL)中并经由套管过滤到小的schlenk中。将该溶液冷冻在-78℃,暴露于动态真空,然后从凉浴中除去以允许苯升华过夜。用-78℃戊烷(2×3mL)洗涤该固体并在真空下干燥过夜,给出53%产率的淡茶色粉末产物(137mg,0.326mmol)。1HNMR(C6D6)δ(ppm):1.64{(2,6)-Me2,6H,s};1.76{Cp(1,3)-Me2,6H,s};1.90{(3,5)-Me2,6H,s};2.03{Cp(2)-Me,3H,s};2.14{(1,7)-Me2,6H,s};4.82{Cp(4,5)-CH}13CNMR(C6D6)δ(ppm):10.5{(2,6)-Me2};11.8{(Cp(2)-Me2};12.1{Cp(1,3)-Me2};12.6{(1,7)-Me2};13.2{(3,5)-Me2};104.2(3,5);105.1{Cp(4,5)};112.1(1,7);118.9{Cp(1,3)};119.3(4);125.1(2,6);126.8(8);127.3{Cp(2)}实施例1c-合成ZrPn*IndCl将[ZrPn*(μ-Cl)3/2]2(μ-Cl)2Li.THF(1.02)(300mg,0.369mmol)溶解在Et2O(20mL)中并冷却到-78℃。经由套管将IndLi(90.1mg,0.738mmol)的浆液转移到该溶液中,并搅拌内容物1h。允许将容器升至室温,然后搅拌另一小时,之后在真空下除去溶剂。将固体少量地再分散于苯(3×2mL)中,并经由套管过滤到小的schlenk中。将溶剂冷冻在-78℃,从冷水浴中除去,并暴露于动态真空过夜。用-78℃戊烷(3×2mL)洗涤产生的粉末,并在真空下干燥4小时,给出75%产率的橙绿色粉末产物(239mg,0.558mmol)。1HNMR(C6D6)δ(ppm):1.49{(2,6)-Me2,6H,s};1.91{(3,5)-Me2,6H,s};1.94{(1,7)-Me2,6H,s};5.51{Ind(2,9),2H,d(3JH-H=3.4Hz)};5.78{Ind(1),1H,t(3JH-H=3.4Hz)};6.93{Ind(5,6),2H,m};7.51{Ind(4,7),2H,m}13CNMR(C6D6)δ(ppm):10.4{(2,6)-Me2};12.5{(3,5)-Me2};13.1{(1,7)-Me2};95.2{Ind(2,9)};105.8(3,5);112.4(1,7);119.1{Ind(1)};119.8(4);123.4{Ind(4,7)};124.0{Ind(5,6)};126.3(2,6);126.5{Ind(3,8);128.6(8)实施例1d-合成Pn*ZrCpnBuCl用玛瑙研磨棒和研钵研磨LiCpnBu(79mg,0.617mmol)并将其添加到包含[Pn*Zr(μ-Cl)3/2]2(μ-Cl)2Li.thf(0.988)(250mg,0.308mmol)的安瓿瓶中。将Et2O(20mL)冷却到-78℃并转移到固体上以及强有力地搅拌1h。将安瓿瓶从冷水浴中除去,并超声1h。然后在室温下搅拌反应混合物另一小时,之后在真空下除去溶剂以提供橙色油,其静置时缓慢结晶。在萃取到苯(3×2mL)中和冷冻干燥之后,作为67%产率的棕色固体(179mg,0.412mmol)给出Pn*ZrCpnBuCl。在-35℃下由饱和的(Me3Si)2O溶液生长适用于X射线衍射研究的单晶体。对于C23H31ClZr的分析计算值(理论值):C,63.63(63.71);H,7.20(7.21).1HNMR(400MHz,C6D6)δ(ppm):0.87(t,3H,3JH-H=7.2Hz,CH3(CH2)3–Cp);1.30(m,2H,CH3CH2(CH2)2-Cp);1.42(m,2H,CH3CH2CH2CH2-Cp);1.70(s,6H,2,6–Me–Pn*);1.84(s,6H,3,5–Me–Pn*);2.11(s,6H,1,7–Me–Pn*);2.6(m,2H,CH3CH2CH2CH2-Cp);5.15(t,2H,3JH-H=2.7Hz,3,4–H–Cp);5.68(t,2H,3JH-H=2.7Hz,2,5–H–Cp).13C{1H}NMR(100MHz,C6D6)δ(ppm):11.3(2,6–Me–Pn*);12.6(1,7–Me–Pn*);13.3(3,5–Me–Pn*);14.3(CH3CH2CH2CH2-Cp);23.0(CH3CH2(CH2)-Cp);29.3(CH3CH2CH2CH2-Cp);33.8(Cp–CH2–CH2–);105.2(3,5–Pn*);106.0(3,4–Cp);112.2(1,7–Pn*);113.4(2,5–Cp);119.5(4–Pn*);125.6(2,6–Pn*);128.6(8–Pn*);130.0(1–Cp).实施例1e-合成Pn*ZrCpMe(Me)将Pn*ZrCpMeCl(200mg,0.510mmol)在甲苯(15mL)中的-78℃溶液添加到MeLi(在Et2O中1.6M,319μL,0.510mmol)在甲苯中的-78℃溶液中。搅拌反应混合物1h,之后暴露于动态真空,同时仍在冷水浴中。从冷水浴中除去溶液,使得溶剂的去除保持在低于0℃的温度。用己烷(4×5mL)萃取固体以及将结合的萃取物浓缩至15mL,以及在冷藏箱中冷却至-80℃过夜,产生黄色微晶固体。除去上清液,以及在真空中干燥固体4h以产生50%产率(95mg,0.256mmol)的Pn*ZrCpMe(Me)。由苯溶液的缓慢蒸发生长适用于X射线衍射研究的单晶体。对于C21H28Zr的分析计算值(理论值):C,67.86(67.73);H,7.59(7.71).1HNMR(400MHz,C6D6)δ(ppm):-0.74(s,3H,Zr-Me);1.63(s,6H,2,6-Me-Pn*);1.85(s,3H,Me-Cp,3H,s);1.99(s,6H,3,5-Me-Pn*);2.00(s,6H,1,7-Me-Pn*);5.15(t,2H,3JH-H=2.6Hz,2,5-H-Cp);5.37(t,2H,3JH-H=2.6Hz,3,4-H-Cp).13C{1H}NMR(C6D6)δ(ppm):10.6(Zr-CH3);10.9(2,6-Me-Pn*);12.4(3,5-Me-Pn*);13.4(1,7-Me-Pn*);14.0(Me-Cp);102.2(1,7-Pn*);105.5(3,4-Cp);106.4(3,5-Pn*);111.4(2,5-Cp);116.9(8-Pn*);119.8(1-Cp);123.1(2,6-Pn*);123.4(4-Pn*).实施例1f-合成Pn*ZrCp(Me)将Pn*ZrCpCl(250mg,0.661mmol)在甲苯(15mL)中的-78℃的溶液添加到MeLi(在Et2O中1.6M,415μL,0.664mmol)在甲苯的-78℃的溶液中。搅拌反应混合物1h,之后暴露于动态真空,同时仍在冷水浴中。从冷水浴中除去溶液,使得溶剂的去除保持在低于0℃的温度。用己烷(4×5mL)萃取固体以及将结合的萃取物浓缩至15mL,以及在冷藏箱中冷却至-80℃过夜,产生黄色微晶固体。除去上清液,以及在真空中干燥固体4h以产生68%产率(160mg,0.447mmol)的Pn*ZrCp(Me)。由苯溶液的缓慢蒸发生长适用于X射线衍射研究的单晶体。1HNMR(400MHz,C6D6)δ(ppm):-0.72(s,1H,Zr-Me);1.64(s,6H,2,6-Me-Pn*);1.96(s,6H,3,5-Me-Pn*);1.97(s,6H,1,7-Me-Pn*);5.46(s,5H,Cp);13C{1H}NMR(C6D6)δ(ppm):10.0(Zr-CH3);11.2(2,6-Me-Pn*);12.3,13.4(3,5-Me-Pn*&1,7-Me-Pn*-由HSQC可区分);102.5(1,7-Pn*);106.4(3,5-Pn*);108.9(Cp);116.9(8-Pn*);123.3(2,6-Pn*);123.6(4-Pn*).实施例1g-合成Pn*ZrCp(H)将Pn*ZrCpCl(150mg,0.397mmol)在甲苯(15mL)中的溶液和三乙基溴氢化钾(在THF中的1.0M溶液,417μL,0.417mmol)填充到安瓿瓶中,并搅拌48h。在真空中除去挥发物,用戊烷(4×5mL)萃取固体并将结合的萃取物浓缩至15mL,之后在冷藏箱中冷却至-80℃过夜。除去上清液,以及在真空中干燥固体4h以产生42%产率(57mg,0.166mmol)的Pn*ZrCp(H)。由苯溶液的缓慢蒸发生长适用于X射线衍射研究的单晶体。1HNMR(400MHz,C6D6)δ(ppm):0.69(d,1H,J=1.6Hz,Zr-H);1.69(s,6H,2,6-Me-Pn*);2.05(s,6H,3,5-Me-Pn*);2.59(s,6H,1,7-Me-Pn*);5.54(s,5H,Cp);13C{1H}NMR(C6D6)δ(ppm):10.8(2,6-Me-Pn*);12.8(3,5-Me-Pn*);14.4(1,7-Me-Pn*);103.9(3,5-Pn*);105.5(Cp);108.6(1,7-Pn*);117.5(4-Pn*);121.4(8-Pn*);121.4(2,6-Pn*).实施例1h-合成Pn*ZrCp(nBu)将Pn*ZrCpCl(200mg,0.529mmol)在甲苯(15mL)中的-78℃的溶液添加到nBuLi(在Et2O中1.6M,347μL,0.555mmol)在甲苯中的-78℃的溶液中。搅拌反应混合物1h,允许缓慢升至室温并搅拌另一小时。然后在动态真空下除去挥发物并用戊烷(4×5mL)提取固体,并将结合的萃取物浓缩至15mL和在冷藏箱冷却至-80℃过夜,产生黄色微晶固体。除去上清液,以及在真空中干燥固体4h以产生57%产率(120mg,0.300mmol)的Pn*ZrCp(nBu)。1HNMR(400MHz,C6D6)δ(ppm):-0.14(m,2H,Zr-CH2-);1.16(t,3H,3JH-H=7.3Hz,Zr-(CH2)3-CH3);1.42(m,2H,Zr-CH2-CH2-);1.58(m,2H,Zr-(CH2)2-CH2-);1.63(s,6H,2,6-Me-Pn*);1.96(s,6H,3,5-Me-Pn*);1.99(s,6H,1,7-Me-Pn*);5.50(s,5H,Cp);13C{1H}NMR(C6D6)δ(ppm):11.0(2,6-Me-Pn*);12.1(3,5-Me-Pn*);13.6(1,7-Me-Pn*);14.6(Zr-(CH2)3-CH3);28.9(Zr-CH2-);32.4(Zr-(CH2)2-CH2-);36.7(Zr-CH2-CH2-);102.4(3,5-Pn*);106.4(1,7-Pn*);108.5(Cp);116.7(4-Pn*);123.1(8-Pn*);123.5(2,6-Pn*).实施例1i-合成Pn*ZrCp(NMe2)在室温下用Pn*ZrCpCl(100mg,0.27mmol)、LiNMe2(19mg,0.37mmol)和苯(20mL)填充安瓿瓶。搅拌生成的黄色溶液3天,过滤并将上清液冷冻在-78℃,以及在动态真空下冻干。作为83%产率(85mg,0.22mmol)的黄色粉末分离Pn*ZrCp(NMe2)。1HNMR(400MHz,C6D6)δ(ppm):1.82(s,6H,2,6-Me-Pn*);1.89(s,6H,3,5-Me-Pn*);2.09(s,6H,1,7-Me-Pn*);2.40(s,6H,N-Me2);5.72(s,5H,Cp)13C{1H}NMR(C6D6)δ(ppm):11.3(2,6-Me-Pn*);12.2(1,7-Me-Pn*);13.8(3,5-Me-Pn*);48.2(N-Me2);104.2(3,5-Pn*);108.0(Cp);112.5(1,7-Pn*);120.3(4-Pn*);125.1(2,6-Pn*);126.7(8-Pn*).实施例1j-合成Pn*ZrCp(NPh2)将THF(20mL)冷却至-78℃并添加到Pn*ZrCpCl(100mg,0.27mmol)和KNPh2.THF0.27(70mg,0.31mmol)中。允许将溶液升至室温,搅拌16h,然后过滤并在动态真空下干燥。将黄色固体再分散于戊烷(20mL)和甲苯(10mL)混合物中,然后过滤。将滤液降至最小体积,并置于-80℃冷藏箱中3天。通过过滤分离生成的黄色晶体,并在真空中干燥以给出60%产率(81mg,0.16mmol)的Pn*ZrCp(NPh2)。1HNMR(400MHz,THFd8)δ(ppm):1.58(s,6H,3,5-Me-Pn*);2.12(s,6H,2,6-Me-Pn*);2.20(s,6H,1,7-Me-Pn*);5.62(s,5H,Cp);6.58(m,6H,N-Phmeta&N-Phpara);7.00(m,4H,N-Phortho).13C{1H}NMR(THFd8)δ(ppm):11.0(3,5-Me-Pn*);11.5(2,6-Me-Pn*);13.6(1,7-Me-Pn*);106.3(1,7-Pn*);111.2(Cp);112.0(3,5-Pn*);117.7(N-Phmeta/para);118.3(8-Pn*);118.3(2,6-Pn*);124.1(N-Phmeta/para)128.3(4-Pn*);128.5(N-Phortho);159.1(N-Phipso).实施例1k-合成ZrPn*FluCl将[ZrPn*(μ-Cl)3/2]2(μ-Cl)2Li.THF(1.02)(51mg,0.060mmol)和LiFlu(21mg,0.12mol)引入到NMR管中并溶解在0.5mL的C6D6中,以及溶液瞬间变为金黄色。将反应混合物加热至80℃持续96h,以及通过C盐过滤苯。除去苯以给出浅绿色固体Pn*ZrFluCl。产率:26mg(80%产率)。1HNMR(苯-d6,400MHz):δ1.251.762.07(s,6H,Pn-CH3),4.69(s,1H,Flu-H),7.04(m,2H,Flu-H),7.08(m,2H,Flu-H),7.17(d,2H,Flu-H,3JHH=7.72Hz),8.31(d,2H,Flu-H,3JHH=8.29Hz).13C{1H}NMR(苯-d6,100MHz):δ9.512.512.8(Pn-CH3),75.5(Flu(C)),118.5(Flu(C)),121.9(Flu(C)),126.5(Flu(C)),127.0(Flu(C)).实施例1l-合成Pn*ZrCpMe(OAm)将Pn*ZrCpMeCl(0.020g,0.051mmol)和KO-2,6-iPr-C6H3(0.006g,0.051mmol)在C6D6(0.5mL)中结合并超声2×30分钟以提供黄色溶液和无色沉淀。使用1HNMR光谱分析溶液表明形成了Pn*ZrCpMe(OAm)。1HNMR(苯-d6,23℃):δ5.705.55(app.t,2Heach,JHH=2.7Hz,C5H4Me),2.07(s,6H,CH3-Pn*),2.02(s,3H,C5H4Me),1.951.89(s,6Heach,CH3-Pn*),1.48(q,2H,3JHH=7.4Hz,C(CH3)2CH2CH3),1.13(s,6H,C(CH3)2CH2CH3),0.86(t,2H,3JHH=7.4Hz,C(CH3)2CH2CH3)、实施例1m-合成Pn*ZrCpMe(O-2,6-Me-C6H3)将Pn*ZrCpMeCl(0.018g,0.046mmol)和KO-2,6-Me-C6H3(0.0090g,0.046mmol)在C6D6(0.5mL)中结合并超声2×30分钟,提供黄色溶液和无色沉淀。随后在真空中干燥滤液以提供浅黄色固体Pn*ZrCpMe(O-2,6-Me-C6H3)。由-30℃的戊烷溶液生长适用于X射线衍射研究的单晶体。1HNMR(苯-d6,23℃):δ7.17(d,2H,3JHH=7.4Hz,3,5-C6H3),6.82(t,1H,3JHH=7.4Hz,4-C6H5),5.475.20(app.t,2Heach,JHH=2.7Hz,C5H4Me),2.081.921.881.84(s,6Heach,CH3-Pn*或2,6-Me-C6H3),1.83(s,3H,C5H4Me).实施例1n-合成Pn*ZrCpMe(O-2,6-iPr-C6H3)将Pn*ZrCpMeCl(0.020g,0.051mmol)和KO-2,6-iPr-C6H3(0.011g,0.051mmol)在C6D6(0.5mL)中结合并超声2×30分钟以提供黄色溶液和无色沉淀。随后在真空中干燥滤液以提供浅黄色固体Pn*ZrCpMe(O-2,6-iPr-C6H3)。1HNMR(苯-d6,23℃):δ7.17(d,2H,3JHH=7.5Hz,3,5-C6H3),6.96(t,1H,3JHH=7.5Hz,4-C6H5),5.635.22(app.t,2Heach,JHH=2.7Hz,C5H4Me),2.93(sept.,2H,3JHH=6.8Hz,CH(CH3)2),2.011.90(s,6Heach,CH3-Pn*),1.89(s,3H,C5H4Me),1.87(s,6H,CH3-Pn*),1.351.21(d,2Heach,3JHH=6.8Hz,CH(CH3)2).实施例1o-合成Pn*ZrCpMe(O-2,4-tBu-C6H3)将Pn*ZrCpMeCl(0.032g,0.082mmol)和KO-2,4-tBu-C6H3(0.020g,0.082mmol)在C6D6(0.5mL)中结合并超声2×30分钟以提供黄色溶液和无色沉淀。随后在真空中干燥滤液以提供浅黄色固体Pn*ZrCpMe(O-2,6-tBu-C6H3)。1HNMR(苯-d6,23℃):δ7.577.26(m,1Heach,3,5,6-C6H3),5.995.665.585.30(m,1Heach,C5H4Me),2.191.991.931.921.901.901.85(s,3Heach,CH3-Pn*或C5H4Me),1.601.43(s,9Heach,O-2,4-tBu-C6H3).实施例1p-合成Pn*ZrCpMe(NMe2)将Pn*ZrCpMeCl(0.045g,0.11mmol)和LiNMe2(0.0058g,0.11mmol)在C6D6(0.5mL)中结合并超声30分钟以提供黄色溶液和无色沉淀。随后在真空中干燥滤液以提供Pn*ZrCpMe(NMe2)作为浅黄色固体。1HNMR(苯-d6,23℃):δ5.665.50(m,2Heach,C5H4Me),2.432.111.92(s,6Heach,CH3-Pn*或NMe2),1.92(s,3H,C5H4Me)1.82(s,6Heach,CH3-Pn*或NMe2).比较例-合成Pn*ZrCptBuCl向-78℃的Et2O(20mL)中的[Pn*Zr(μ-Cl)3/2]2(μ-Cl)2Li.Et2O(1.21)(300mg,0.362mmol)中添加LiCptBu在Et2O(15mL)中的-78℃浆液。在1h的过程中将反应混合物升至室温,然后搅拌1h。在真空下除去挥发物,以及将固体萃取到苯(3×2mL)中并冻干。用-78℃戊烷(2×3mL)洗涤固体并在真空下干燥4h以提供80%产率(253mg,0.583mmol)的Pn*ZrCptBuCl。通过由-78℃的戊烷再结晶产物制备分析样品。由苯溶液的缓慢蒸发生长适用于X射线衍射研究的单晶体。对于C23H31ClZr的分析计算值(理论值):C,63.63(63.55);H,7.20(7.33).1HNMR(400MHz,C6D6)δ(ppm):1.33(s,9H,tBu-Cp);1.71(s,6H,2,6-Me-Pn*);1.83(s,6H,3,5-Me-Pn*);2.09(s,6H,1,7-Me-Pn*);5.04(t,2H,3JH-H=2.8Hz,2,5-H-Cp);5.96(t,2H,3JH-H=2.8Hz,3,4-H-Cp).13C{1H}NMR(100MHz,C6D6)δ(ppm):11.6(2,6-Me-Pn*);12.6(1,7-Me-Pn*);13.3(3,5-Me-Pn*);32.2(CMe3-Cp);104.2(2,5-Cp);105.0(3,5-Pn*);112.3(1,7-Pn*);113.9(3,4-Cp);119.4(4-Pn*);126.1(2,6-Pn*);128.6(8-Pn*);138.8(1-Cp).实施例2-合成催化组合物实施例2a-将前体催化剂支撑在聚甲基铝氧烷上根据以下通用方法将实施例1a-c的化合物支撑在固体MAO上。在室温下将甲苯(40ml)添加到包含固体铝氧烷(300mg,5.172mmol)(固体MAO)(由TOSOH生产,批号TY130408)和Pn*ZrCpMeCl(10.1mg,0.0258mmol)(以上所示)的Schlenk管中。将浆液加热到80℃,然后在偶尔的打旋下放置两小时,其间溶液变为无色,以及固体上色。然后将产生的悬浮液冷却至室温,以及在真空中仔细过滤甲苯溶剂并将其除去以得到作为自由流动的粉末的固体MAO/Pn*ZrCpMeCl催化剂,产率88%(273mg)。实施例2b-用于将前体催化剂支撑在固体聚甲基铝氧烷上的替换方法根据以下通用方法将实施例1a-j的化合物支撑在固体聚甲基铝氧烷上。将聚甲基铝氧烷与前体催化剂组合并一起搅拌干燥5分钟。停止搅拌,以及将甲苯(10mL)添加到混合物中,并加热至60℃持续1h。每5分钟手动打旋内容物,以及1h之后允许沉淀,留下有色固体和无色溶液。经由套管除去上清液并在真空下干燥固体4h。产生的支撑的前体催化剂的特征如下:Pn*ZrCpCl-sMAO13CCPMAS(10KHz)δ(ppm):-13.3(sMAO-Me);6.3(Pn*-Me);106.2(Cp-环;Pn*-环);123.0(sMAO-苯甲酸盐);126.4(sMAO-苯甲酸盐);172.0(sMAO-苯甲酸盐α-C(O)O).27AlDPMAS(15KHz)δ(ppm):-387.4;-250.7;-83.1;75.7;211.2;347.0.Pn*ZrCpMeCl-sMAO13CCPMAS(10KHz)δ(ppm):-13.3(sMAO-Me);6.7(Pn*-Me);15.1(Cp-Me);106.7(Cp-环;Pn*-环);122.9(sMAO-苯甲酸盐);126.0(sMAO-苯甲酸盐);130.8(sMAO-苯甲酸盐);172.4(sMAO-苯甲酸盐α-C(O)O).27AlHAHNECHO(15KHz)δ(ppm):-242.4;-86.4;97.6;179.2;330.3.Pn*ZrCptBuCl-sMAO13CCPMAS(10KHz)δ(ppm):-13.3(sMAO-Me);6.7(Pn*-Me);26.9(Cp-C-Me3);111.2(Cp-环;Pn*-环);123.0(sMAO-苯甲酸盐);126.0(sMAO-苯甲酸盐);130.7(sMAO-苯甲酸盐);171.9(sMAO-苯甲酸盐α-C(O)O).27AlDPMAS(15KHz)δ(ppm):-246.8;-80.5;74.2;204.4;351.0.Pn*ZrCpnBuCl-sMAO13CCPMAS(10KHz)δ(ppm):-13.3(sMAO-Me);6.5(Pn*-Me);15.9(Cp-正丁基);23.0(Cp-正丁基);28.4(Cp-正丁基);108.6(Cp-环;Pn*-环);123.1(sMAO-苯甲酸盐);126.2(sMAO-苯甲酸盐);131.2(sMAO-苯甲酸盐);172.5(sMAO-苯甲酸盐α-C(O)O).27AlHAHNECHO(15KHz)δ(ppm):-241.1;-84.2;72.4;204.3;343.2.Pn*ZrCpMe3Cl-sMAO13CCPMAS(10KHz)δ(ppm):-13.4(sMAO-Me);6.2(Pn*-Me);107.2(Cp-环;Pn*-环);123.0(sMAO-苯甲酸盐);126.0(sMAO-苯甲酸盐);130.7(sMAO-苯甲酸盐);172.0(sMAO-苯甲酸盐α-C(O)O).27AlDPMAS(15KHz)δ(ppm):-385.1;-247.7;-78.1;32.7;203.7;349.0.Pn*ZrIndCl-sMAO13CCPMAS(10KHz)δ(ppm):-13.3(sMAO-Me);6.3(Pn*-Me);122.9(sMAO-苯甲酸盐);130.8(sMAO-苯甲酸盐);172.8(sMAO-苯甲酸盐α-C(O)O).27AlDPMAS(15KHz)δ(ppm):-382.5;247.1;-75.2;33.2;209.4;353.0.Pn*ZrCpMeMe-sMAO13CCPMAS(10KHz)δ(ppm):-13.3(sMAO-Me);6.2(Pn*-Me);106.4(Cp-环;Pn*-环);122.8(sMAO-苯甲酸盐);126.0(sMAO-苯甲酸盐);130.0(sMAO-苯甲酸盐);171.7(sMAO-苯甲酸盐α-C(O)O).27AlHAHNECHO(15KHz)δ(ppm):-240.0;-81.1;83.0;208.6;345.9.实施例3-聚合研究实施例3a-用于乙烯的浆液相聚合的一般程序用TiBA(150mg,0.756mmol)、甲苯(50mL)和支撑的催化剂(10mg)填充安瓿瓶。将内容物放置在要求温度的油浴中并允许平衡5分钟,同时脱除顶部空间的空气。向烧瓶通乙烯(2巴),并在1200rpm下搅拌整个实验期间。然后过滤聚合物,用戊烷(2×20mL)洗涤,以及在5毫巴下干燥过夜。实施例3b-乙烯聚合活性(固体MAO支撑的浆液相)使四种锆复合物与固体MAO反应,并在以下条件下用于乙烯的固体MAO支撑的浆液聚合:1:200的[M]:[TIBA],50mL甲苯,60℃和30分钟。图2和表1中示出了结果。表1.各种Pn*ZrCpRCl复合物的固体MAO浆液聚合的总结实施例3c-乙烯聚合活性(固体MAO支撑的浆液相)按照实施例3a中列出的流程,评估各种MAO支撑的前体催化剂在乙烯聚合中的催化活性。表2和图3中列出了结果。表2.使用各种Pn*ZrCpRCl(CpR=Cp、CpMe、CptBu、CpnBu、CpMe3、Ind)和Pn*ZrCpMeMe的固体MAO浆液聚合的总结。聚合条件:[Zr]:[MAO]=1:250;150mgTiBA助催化剂;2巴乙烯;10mg催化剂负荷;50mL甲苯;60℃;30分钟。复合物温度(℃)时间(分钟)活性(kgPE/(molZr·h·巴))Pn*ZrCpCl60303624±256Pn*ZrCpMeCl60304209±73Pn*ZrCptBuCl6030294±86Pn*ZrCpnBuCl60304096±72Pn*ZrCpMe3Cl60301981±507Pn*ZrIndCl60301971±77Pn*ZrCpMe(Me)60304486±300表2示出了通过CpR的变化可以观察到比值的显著改进。实施例3d-聚乙烯特征确定使用各种固体MAO支撑的前体催化剂制备的聚乙烯的分子量(Mw和Mn)和多分散指数(PDI)。表3和图4中列出了结果。表3.在60℃下在Pn*ZrCpRCl(CpR=Cp、CpMe、CptBu、CpMe3、Ind)的情况下聚甲基铝氧烷支撑的乙烯聚合的聚合物分子量Mw。在括号中给出了PDI。聚合条件:[Zr]:[sMAO]=1:200;150mgTiBA助催化剂;2巴乙烯;10mg催化剂负荷;50mL甲苯;60℃;30分钟复合物温度(℃)时间(分钟)Mw(Kg/mol)Mn(Kg/mol)PDIPn*ZrCpCl60303250001370002.4Pn*ZrCpMeCl60302500001030002.4Pn*ZrCptBuCl6030310000440007Pn*ZrCpMe3Cl60305050001330003.8Pn*ZrIndCl6030290000860003.4通过GPC确定使用固体-MAO支撑的Pn*ZrCptBuCl制备的聚乙烯的分子量分布。该结果在图5中示出。表3示出了通过CpR的变化可以控制分子量。实施例3e-温度对固体-MAO支撑的Pn*ZrCpMeCl的催化活性的影响。评估在固体MAO支撑的Pn*ZrCpMeCl的情况下的乙烯聚合活性的温度依赖性。表4和图6中列出了结果。表4.在固体MAO支撑的Pn*ZrCpMeCl的情况下的乙烯聚合活性的温度依赖性。聚合条件:[Zr]:[sMAO]=1:200;150mgTiBA助催化剂;2巴乙烯;10mg催化剂活性;50mL甲苯;30分钟。复合物温度(℃)时间(分钟)活性(kgPE/(molZr·h·巴))Pn*ZrCpMeCl40302563±103Pn*ZrCpMeCl50302988±34Pn*ZrCpMeCl60304209±73Pn*ZrCpMeCl70303368±164Pn*ZrCpMeCl80302841±62表4示出了对于Pn*ZrCpMeCl,在60℃观察到最大活性。实施例3f-温度对固体-MAO支撑的Pn*ZrCpnBuCl的催化活性的影响。评估在固体MAO支撑的Pn*ZrCpnBuCl的情况下的乙烯聚合活性的温度依赖性。表5和图7中列出了结果。表5.在固体MAO支撑的Pn*ZrCpnBuCl的情况下乙烯聚合活性的温度依赖性。聚合条件:[Zr]:[sMAO]=1:200;150mgTiBA助催化剂;2巴乙烯;10mg催化剂负荷;50mL甲苯;30分钟。复合物温度(℃)时间(分钟)活性(kgPE/(molZr·h·巴))Pn*ZrCpnBuCl40304786±156Pn*ZrCpnBuCl50305025±213Pn*ZrCpnBuCl60304096±72Pn*ZrCpnBuCl70303446±195Pn*ZrCpnBuCl80303502±62表5示出了对于Pn*ZrCpnBuCl,在50℃观察到最大活性。实施例3g-温度对固体-MAO支撑的Pn*ZrCpMe(Me)的催化活性的影响。评估在固体MAO支撑的Pn*ZrCpMe(Me)的情况下的乙烯聚合活性的温度依赖性。表6和图8中列出了结果。表6.在固体MAO支撑的Pn*ZrCpMe(Me)的情况下乙烯聚合活性的温度依赖性。聚合条件:[Zr]:[sMAO]=1:200;150mgTiBA助催化剂;2巴乙烯;10mg催化剂负荷;50mL甲苯;30分钟。复合物温度(℃)时间(分钟)活性(kgPE/(molZr·h·巴))Pn*ZrCpMe(Me)40302644±252Pn*ZrCpMe(Me)50305012±50Pn*ZrCpMe(Me)60304376±441Pn*ZrCpMe(Me)70303074±66Pn*ZrCpMe(Me)80302968±70表6示出了对于Pn*ZrCpMe(Me),在50℃观察到最大活性。实施例3h-反应持续时间对固体-MAO支撑的Pn*ZrCpCl的催化活性和产生的聚乙烯的特征的影响评估固体MAO-支撑的Pn*ZrCpCl的情况下乙烯聚合活性和聚合物分子量(MW)的时间依赖性。表7和图9中列出了结果。表7.在聚甲基铝氧烷支撑的前体催化剂Pn*ZrCpCl的情况下乙烯聚合活性和聚合物分子量MW的时间依赖性。还给出了PDI。聚合条件:[Zr]:[sMAO]=1:200;150mgTiBA助催化剂;2巴乙烯;10mg催化剂负荷;50mL甲苯;60℃。表7示出了通过改变聚合的持续时间可以控制聚乙烯的分子量,同时观察对活性的影响。实施例3i-温度对固体-MAO支撑的Pn*ZrCpMe(Me)的催化活性和产生的聚乙烯的特征的影响评估在固体MAO-支撑的Pn*ZrCpCl的情况下乙烯聚合活性和聚合物分子量(MW)的温度依赖性。表8和图10中列出了结果。表8.在聚甲基铝氧烷支撑的前体催化剂Pn*ZrCpCl的情况下乙烯聚合活性和聚合物分子量MW的温度依赖性。还给出了PDI。聚合条件:[Zr]:[sMAO]=1:200;150mgTiBA助催化剂;2巴乙烯;10mg催化剂负荷;50mL甲苯。表8示出了通过改变聚合的持续时间可以控制聚乙烯的分子量,同时观察对活性的影响。实施例3j-温度对固体-MAO支撑的Pn*ZrCp(NMe2)的催化活性的影响评估在固体MAO支撑的Pn*ZrCp(NMe2)的情况下的乙烯聚合活性的温度依赖性。表9和图11中列出了结果。表9.在固体MAO支撑的Pn*ZrCp(NMe2)的情况下乙烯聚合活性的温度依赖性。聚合条件:[Zr]:[sMAO]=1:200;150mgTiBA助催化剂;2巴乙烯;10mg催化剂负荷;50mL甲苯;30分钟。复合物温度(℃)时间(分钟)活性(kgPE/(molZr·h·巴))Pn*ZrCp(NMe2)40302905±83Pn*ZrCp(NMe2)50302426±195Pn*ZrCp(NMe2)60302337±86Pn*ZrCp(NMe2)70302197±5Pn*ZrCp(NMe2)80301645±27表9示出了与其中Y=Cl或甲基的组合物相比,当Y=NMe2时,在40℃观察到最大活性。实施例3k-温度对固体-MAO支撑的Pn*ZrCp(NPh2)的催化活性的影响评估在固体MAO支撑的Pn*ZrCp(NPh2)的情况下的乙烯聚合活性的温度依赖性。表10和图12中列出了结果。表10.在固体MAO支撑的Pn*ZrCp(NPh2)的情况下乙烯聚合活性的温度依赖性。聚合条件:[Zr]:[sMAO]=1:200;150mgTiBA助催化剂;2巴乙烯;10mg催化剂负荷;50mL甲苯;30分钟。复合物温度(℃)时间(分钟)活性(kgPE/(molZr·h·巴))Pn*ZrCp(NPh2)40302361±291Pn*ZrCp(NPh2)50301945±212Pn*ZrCp(NPh2)60301715±366Pn*ZrCp(NPh2)70301332±301Pn*ZrCp(NPh2)80301090±384表10示出了与其中Y=Cl或甲基的组合物相比,当Y=NPh2时,在40℃观察到最大活性。实施例3l-反应持续时间对固体-MAO支撑的Pn*ZrCp(NMe2)的催化活性的影响评估在固体MAO支撑的Pn*ZrCp(NMe2)的情况下的乙烯聚合活性的时间依赖性。表11和图13中列出了结果。表11.在聚甲基铝氧烷支撑的前体催化剂Pn*ZrCp(NMe2)的情况下乙烯聚合活性的时间依赖性。聚合条件:[Zr]:[sMAO]=1:200;150mgTiBA助催化剂;2巴乙烯;10mg催化剂负荷;50mL甲苯;40℃。复合物温度(℃)时间(分钟)活性(kgPE/(molZr·h·巴))Pn*ZrCp(NMe2)4055989±63Pn*ZrCp(NMe2)40153786±159Pn*ZrCp(NMe2)40302949±105Pn*ZrCp(NMe2)40602350±15表11表明活性随着聚合时间增加而降低。实施例3m-反应持续时间对固体-MAO支撑的Pn*ZrCp(NPh2)的催化活性的影响评估在固体MAO支撑的Pn*ZrCp(NPh2)的情况下的乙烯聚合活性的时间依赖性。表12和图14中列出了结果。表12.在聚甲基铝氧烷支撑的前体催化剂Pn*ZrCp(NPh2)的情况下乙烯聚合活性的时间依赖性。聚合条件:[Zr]:[sMAO]=1:200;150mgTiBA助催化剂;2巴乙烯;10mg催化剂负荷;50mL甲苯;40℃。复合物温度(℃)时间(分钟)活性(kgPE/(molZr·h·巴))Pn*ZrCp(NPh2)4054213±95Pn*ZrCp(NPh2)40152618±11Pn*ZrCp(NPh2)40302361±147Pn*ZrCp(NPh2)40601718±37表12表明活性随着聚合时间增加而降低。实施例3n-形态学研究图15示出了使用固体-MAO支撑的Pn*ZrCpMeCl、Pn*ZrIndCl和Pn*ZrCptBuCl(比较)通过乙烯的浆液相聚合制备的聚乙烯的形态。图15示出了可以将本发明的组合物用于制备具有均匀的形态的聚乙烯。虽然出于参照和示出的目的,本文已经描述了本发明的具体实施方式,但是在没有背离所附权利要求所定义的本发明的范围的情况下,各种修改对于本领域所属技术人员来说是显而易见的。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1