一种亚乙基脲的合成方法与流程
本发明涉及一种化学中间体的合成方法,特别是一种尿素法生产亚乙基脲的方法。
背景技术:
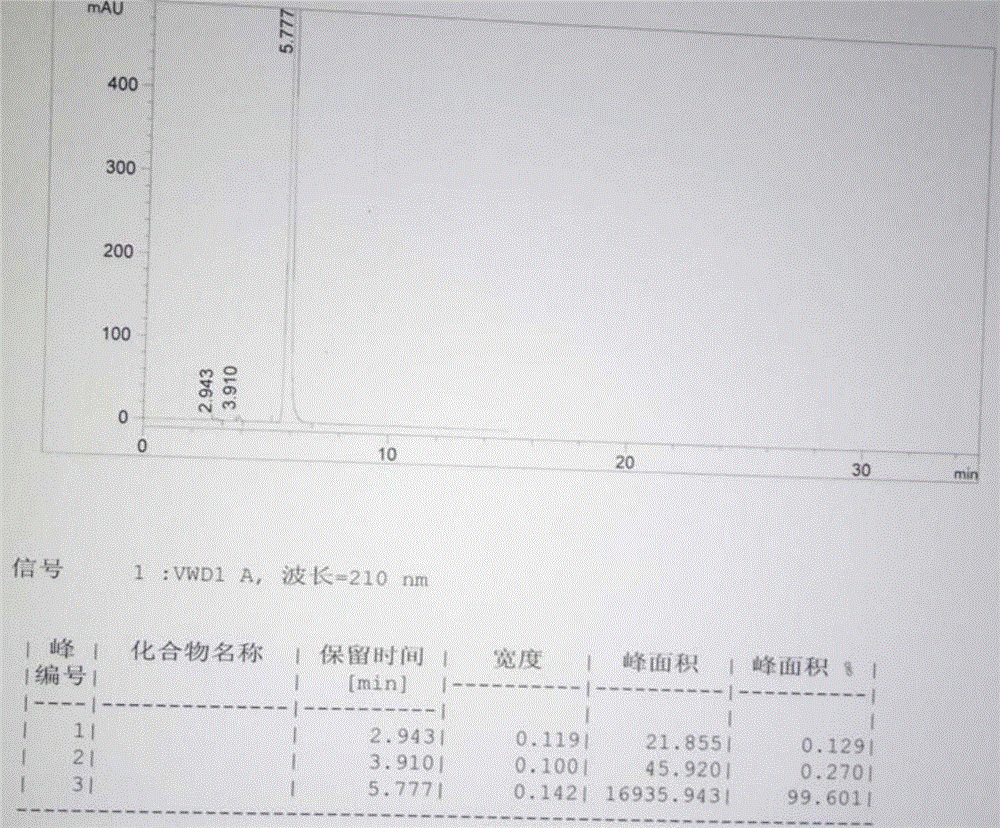
:亚乙基脲,别名:乙烯脲,2-咪唑烷酮,环亚乙基脲。在医药行业中,亚乙基脲为多种新型抗生素,如美洛西林、阿洛西林的关键中间体,并作为第三代青霉素的基础原料,也可以用做抗血吸虫病药的中间体,另外该产品对于除甲醛有奇特的功效,并且能有效的去除经2D-树脂、KB树脂、脲醛树脂、三聚氰胺甲醛树脂等整理后织物中残存的甲醛,因此也可用于合成高性能甲醛清除剂、家居空气甲醛强效活力酶等,在生物领域,它可生产植物生长调节剂、杀菌剂、抑制剂、除草剂等。在本发明之前,合成亚乙基脲的技术方法主要是光气法、固体光气法和尿素法。光气法和固体光气法存在安全、污染环境等隐患;现有尿素法是以尿素和乙二胺为原料,将乙二胺与尿素、助剂按比例加入反应瓶中,慢慢升温至沸腾,并在继续升温的情况下慢慢回收助剂和过量的乙二胺,直至反应温度达到250℃,控制反应过程5小时,并在250℃保温2小时,冷却后得到固体混合物,对固体混合物重结晶,得亚乙基脲;尿素法合成亚乙基脲时会产生聚合物杂质,由于聚合物会随亚乙基脲一并析出,重结晶无法将聚合物除去;为了克服上述尿素法生产亚乙基脲的缺陷,本发明人曾在中国专利CN102030711A中提出了蒸发并收集得到了亚乙基脲的技术方案,这个方案虽然克服了如上的缺点,但仍存在能耗高的问题,并且升华产物含量较低,需要进一步重结晶才能得到高含量的目标产物,不利于降低生产成本。技术实现要素:本发明的目的主要解决现有尿素法生产亚乙基脲的技术缺点,提供一种工艺合理、反应收率高、生产成本低、环保友好的亚乙基脲的合成方法。本发明的目的由以下技术方案实现:一种亚乙基脲的合成方法,其特征在于:在反应器内,加入至少乙二胺和尿素两种原料,然后加热,随着温度的升高,尿素和乙二胺进行缩合反应并产生氨气,反应至氨气的产生明显减少或停止逸出,然后降温并加入溶剂,使其中亚乙基脲呈结晶状态析出,溶剂中的聚合物杂质呈固体悬浮状态析出,对含有呈固体悬浮状态的聚合物杂质的液体部分进行过滤,除去聚合物杂质,然后再分离出反应器内的结晶物料,干燥后得目标产物亚乙基脲。本发明的目的还可以下述技术方案实现:所述的溶剂为水、甲醇、乙醇、丙醇、丙酮、乙腈、氯仿、二氯甲烷中的一种或二种以上混合物。在加入乙二胺和尿素两种原料的同时,还加入助剂水和乙二醇。通过使反应器内处于负压状态或在反应器内通入惰性气体的方法促使氨气的快速逸出。在通过过滤方式除去呈固体悬浮状态的聚合物杂质后,反应器内的结晶物料采用直接过滤方式分离,或者再次加热溶解结晶物料,然后通过再次结晶后第二次分离,干燥后获得目标产物亚乙基脲。第一批分离出目标产物亚乙基脲后的母液直接套用于下一个批次的生产,或者回收溶剂后套用于下一个批次的生产;如此循环若干个批次,最后一批次分离出目标产物亚乙基脲后的母液,通过浓缩后,对浓缩液先过滤除去呈固体悬浮状态的聚合物杂质后,然后再分离得到结晶目标产物。本发明具有如下有益效果:采用本发明方法第一遍即可得到含量在99%以上的高质量产品,生产工艺合理,投资低,能耗低且能够做到杂质每批清,有利于质量管控。附图说明图1利用加热蒸发方法收集的产物液相图谱图2利用加热蒸发方法对收集的产物进行重结晶后的液相图谱图3按照本发明方法一次分离出的产品的液相色谱图(实施例1)图4按照本发明方法一次分离出的产品的液相色谱图(实施例2)图5现有技术康壮产品液相色谱图图6现有技术万福多产品液相色谱图图7本发明产品与现有技术产品比色管目视比色法的浊度对比图具体实施方式本发明用以下实施例对本技术方案做非限制性的进一步说明。本发明在申请人已作出“一种2-咪唑烷酮的合成方法”(CN102030711A)的基础上进一步研究发明的。CN102030711A的技术方案也是以尿素和乙二胺为原料加温进行反应,收集初馏分、目标产物和残留物分别占本批投料理论产量的5%、70%、25%,初馏分和残留物套用到下一批,得到了2-咪唑烷酮产品。上述方法存在的主要问题是,通过对反应物加热蒸发,然后分段收集产物,使杂质留在反应器内蒸发收集的产品含量低,在94%左右,需要二遍精制才能达到99%。由于2-咪唑烷酮沸点很高高,蒸发收集产物要消耗大量的能量,能耗高。总有约30%的物料留在反应罐内循环,无法做到批清,不利于质量管控需要真空、收集设施,设备要求精度高,投资高。本发明人在选择溶剂时观察溶液浊度时偶尔发现:取一定量的反应混合物在乙醇中加热溶解,然后放置,在放置过程中,发现2-咪唑烷酮以较大的粒状结晶析出,而杂质能够悬浮在乙醇中,结果是2-咪唑烷酮和杂质在乙醇中是以完全不同的两种状态存在,可以用一定目数的滤网将2-咪唑烷酮和液体阻隔开,使悬浮有杂质的液体通过反复过滤将杂质分离出来。进一步研究发现,除乙醇单独可以做溶剂用于分离外,乙醇、水、甲醇、丙醇、丙酮、乙腈、氯仿、二氯甲烷中的一种或二种以上混合物,做溶剂进行结晶现象观察试验,方法同上,发现所选溶剂均能够使2-咪唑烷酮以较大的粒状结晶析出,而杂质能够悬浮在溶剂中,因此可以用于上述的反应物与杂质的分离。优先采用乙醇,主要是考虑乙醇毒性低,汽化潜热低,在工业应用比较广泛。在反应罐中加入助剂,主要有水和乙二醇,水和乙二醇主要有两个作用,一是水和乙二醇能够促使尿素和乙二胺互溶,促进加快反应,二是减少反应过程中局部类似爆沸的现象,反应过程不加上述两种物质也可以,但反应时间长一些,而且在160℃附近时,由于大量氨气的产生,易使罐内物料呈现严重发泡状,粘稠,而不易控制。当采用一次性制备目标产物亚乙基脲时,由于第一次结晶是在含有大量杂质的溶液中析出的,结晶颗粒总会有一部分发生粘连,结果是会有微量的杂质夹杂在粘连的结晶中间;将溶液中的杂质反复过滤除去后,溶液中的杂质与起初相比大为减少,此时加热溶解再结晶,相当于进行了一次重结晶,有利于减少粘连结晶夹杂的杂质。当采用多批次循环方法制备目标产物亚乙基脲时,母液中溶解一定量的2-咪唑烷酮,母液直接套用,可以实现在反应升温过程中直接回收溶剂,减少流程,并同时完成了其中的2-咪唑烷酮留在了反应器内,从工艺上这相对生产是合理的。这里所说的批清是相对而言,相对于CN102030711A方法,本工艺的杂质每一批都从罐内直接剔除掉了,产品含量相对平稳,而CN102030711A方法中的杂质一直在罐内残存,结果是中间馏份的含量呈逐渐下降态势;相对于CN102030711A,本发明每一批料可很直观的看到料全部从管内放出来,而CN102030711A方法在实验室的玻璃罐内还好控制,在实际生产中初馏分、目标产物、残留物只能按比例大体估算,所以造成实际生产批量波动较大。关于本发明中的下述概念的含义如下:收率:本发明的化学反应中乙二胺过量,收率是按照尿素折纯后为基准计算的,计算公式:产品实际产量÷(50×86÷60)×100%=收率含量和溶液浊度:2-咪唑烷酮作为中间体化工产品使用,目前没用行业标准,都是企业自定标准,主要参考指标为含量、熔点和水溶液浊度。含量分析作为企业秘密不尽相同,有的是仪器法,也有的采用化学法;水溶液浊度是取1g样品,加纯水稀释至10ml使其溶解,观察溶液的澄清程度以客户能够接受为依据,目前所有企业均不能提供浊度数据;熔点即常规检测。由于没有通用的方法参考,本发明人对含量的分析采用的是化学法:取一定量样品,用纯净水溶解,加入过量硫酸标准溶液,加热回流30分钟,改为蒸馏,蒸出约一半的水量,降温到室温,用与蒸出水量相当的新纯净水冲洗瓶口和罐壁,以甲基红作指示剂,用氢氧化钠标准溶液返滴定过量的硫酸,算出消耗的硫酸量,同时用标准品做平衡对比算出样品含量。标准品是自己做的,试验样品用无水乙醇重结晶3遍,用液相色谱检测无杂质峰作为标准。溶液浊度:是用浊度计(上海昕瑞仪器)检测的,用400NTU浊度标准液稀释成40浊度标准液,使用前用40浊度标准试样放仪器内调40,用空瓶调零点,样品配成10g/100ml的水溶液,测浊度。实施例1:在装有回流冷凝管、搅拌器、温度计的250ml的反应器内,加入51.4g尿素(含量97.27%),80ml乙二胺,折合后纯尿素与乙二胺分子比为1:1.44,30ml水,30ml乙二醇;加热升温,随着温度的升高尿素逐渐溶解,继续升温,控制加温速度使温度均匀上升并开始回收物料,回收的物料为水、乙二醇、乙二胺混合物,直接套用于下一批,在6小时温度逐渐上升到240℃时,控制在240~260℃保温1h,停止回收,共回收物料78ml;降温到90℃,加入100ml乙醇,再降温到30℃,静置沉降;对悬浮有固体杂质的液体部分过滤剔除悬浮杂质,然后分离出反应罐内的结晶物料:反应罐内的物料用布氏漏斗抽滤,用50ml乙醇分两次洗涤,共得母液163ml,母液套用于下一批,结晶物料干燥后得目标产物41.8g,收率58.3%,含量99.4%,检测目标产物水溶液浊度1.33。说明:使用的尿素为工业级,乙二胺、乙二醇、乙醇为分析纯,以下实施例均相同。实施例2:在装有回流冷凝管的、搅拌器、温度计、恒压漏斗的250ml的反应器内,加入51.4g尿素(含量97.27%)、56ml乙二胺(折合后纯尿素与乙二胺分子比为1:1)、水5ml,加入实施例1的母液80ml,再在恒压漏斗中加入实施例1的母液83ml和回收物料76ml的混合液,然后加热回收乙醇,并开始滴加回收物料和母液的混合液,保持滴加的速度不至于罐满,直至全部加入,继续回收至罐内温度达到100℃的部分为乙醇和水的混合物,共回收138ml(用于本批溶解和洗涤),并继续加热回收物料,控制加温速度使温度均匀上升,在6小时温度逐渐上升到240℃时,控制在240~260℃保温1h,停止回收,共回收物料79ml(用于下一批套用),降温到90℃,加入100ml回收乙醇,加2g活性炭,控制在60℃,搅拌30分钟,过滤除去活性炭,滤液降温到30℃,静置沉降,对悬浮有固体杂质的液体部分过滤剔除悬浮杂质,然后分离出反应罐内的结晶物料:反应罐内的物料用布氏漏斗抽滤,用50ml乙醇分两次洗涤(用本批回收乙醇38ml,加12ml新鲜乙醇),共得母液161ml(母液套用于下一批),固体干燥得目标产物72.8g,收率101.6%,含量99.7%,检测目标产物水溶液浊度1.21。采用实施例2的操作方法,用母液和回收物料循环生产到第10批,实施例3~10的投料量均为51.4g尿素、56ml乙二胺,不再投水。目标产物结果如下:批号12345678910含量(%)99.499.799.398.999.299.699.498.699.399.4收率(%)58.3101.697.699.298.5100.598.297.5100.298.6水溶液浊度1.331.211.291.521.481.361.411.621.301.35实施例11:对实施例10的母液浓缩回收乙醇,至罐内温度85℃,降温到65℃,加入2g活性炭,搅拌30分钟,过滤,滤液降温到10℃,对悬浮有固体杂质的液体部分过滤剔除其中的悬浮杂质,然后分离出反应罐内的结晶物料,干燥得目标产物21.3g,目标产物含量99.1%水溶液浊度2.31。以上实施例1-11是循环使用上批母液和回收物料方式的生产方法的整个操作步骤。实施例12:在装有回流冷凝管的250ml的反应器内,加入51.4g尿素(含量97.27%)、60ml乙二胺(折合后纯尿素与乙二胺摩尔比1:1.08),加热升温,随着温度的升高尿素逐渐溶解,继续升温,控制加温速度使温度均匀上升,在8~9小时温度逐渐上升到240℃时,控制在240~260℃保温1h,降温到90℃,加入100ml乙醇,降温到20℃,静置沉降,对悬浮有固体杂质的液体部分过滤剔除其中的悬浮杂质,然后加热至70℃,使罐内物料全部溶解,再用2小时降温到10℃析结晶,反应罐内的物料用布氏漏斗抽滤,用50ml乙醇分两次洗涤,共得母液152ml,固体干燥后得目标产物62.7g,收率87.4%,含量99.4%,检测目标产物水溶液浊度1.27。实施例13:相同条件重复实施例12,共得母液150ml,干燥后得目标产物62.06g,收率86.6%,含量99.32%,检测目标产物水溶液浊度1.35。实施例14:对实施例12和实施例13的母液合并浓缩回收乙醇,至罐内温度83℃,降温到65℃,加入2g活性炭,搅拌30分钟,过滤,滤液降温到10℃,对悬浮有固体杂质的液体部分过滤剔除其中的悬浮杂质,然后分离出反应罐内的结晶物料,干燥得目标产物16.4g,目标产物含量98.9%,水溶液浊度2.46。用同样的方法检测国内某企业固体光气法生产的产品的含量和水溶液浊度为:含量99.2%,浊度2.03。本发明亚乙基脲的合成方法与申请人在先已获专利“一种2-咪唑烷酮的合成方法”(CN102030711A)的实质性区别与优缺点对比列表如下:本发明的产品质量及与市场销售产品质量性能比较参见图3-图7。1,为了对照产品水溶液浊度,从市场购买了河北沧州康壮化工公司(以下简称康壮)和济南万福多化工公司的产品进行比较,采用了比色管目视比色法和使用浊度计检测两种方法,本产品明显比两者澄清度要好。图7是比色管目视比色法的图片,方法是各取1g样品,加水溶解并稀释到10ml,然后对比浊度。2,检测产品的熔点:3、液相色谱图比较:参见图3-图6,从色谱图上的相对含量看,康壮和万福多的杂质明显高于本产品,其中万福多产品杂质十分多。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1