N,N‑二烷基取代吡唑离子液体、制备方法及其催化合成碳酸丙烯酯的方法与流程
本发明属于有机合成技术领域,具体涉及一种新型N,N-二烷基取代吡唑离子液体、制备方法,同时还涉及一种利用其催化合成碳酸丙烯酯的方法。
背景技术:
离子液体是完全由离子组成的一类特殊的液体熔融盐,近年来,离子液体作为一种清洁溶剂和新型催化材料得到催化界及石化企业的青睐。在种类繁多的离子液体中,吡唑离子液体的研究虽然不是最多,但其展现的优良性质越来越受到人们的关注。通过改变阴阳离子的不同组合,可以合成多种类型的吡唑离子液体。目前,已有一些吡唑离子液体被设计与合成出来,并被应用于电化学、有机催化反应、有机功能材料和表面活性剂等领域。
合成离子液体的方法基本有两种:一种是复分解法,另一种是酸碱中和法。至今离子液体尚未实现大规模的工业化生产,主要由于离子液体造价昂贵且提纯困难。Namboodiri等人研究了用微波技术促进的非溶剂法合成1,3-二烷基卤化铵和1,3-二烷基四氟硼酸盐室温离子液体的方法,然而微波法的反应条件不易控制,而且这种方法要求特殊设备,不利于大规模工业化推广利用。
在庞大的离子液体体系中,咪唑类和吡啶类离子液体是合成最多的,虽然也有部分的吡唑离子液体得到合成,但对于吡唑离子液体的研究相对较少,对其性质的了解也很少。
技术实现要素:
本发明目的在于克服现有技术缺陷,提供一种新型的N,N-二烷基取代吡唑离子液体、制备方法以及利用其催化合成碳酸丙烯酯的方法。
为实现上述目的,本发明采用如下技术方案:
一种N,N-二烷基取代吡唑离子液体,其结构通式如下:
;
式中,R为CH3,n为2、4、5、7、9或11;X为Br、I、BF4或PF6。
上述N,N-二烷基取代吡唑离子液体的制备方法,其包括如下步骤:
1)将吡唑与粉末状KOH在乙腈溶剂中常温搅拌1~2h;
2)向步骤1)所得体系中加入卤代烷,并于50~120℃反应16~36h;
3)反应结束后,浓缩溶剂,经后处理即得。后处理具体可以是:加入CH2Cl2、过滤,滤液浓缩后,再用乙酸乙酯洗涤;析出固体,过滤,即得相应的吡唑离子液体产物。真空干燥后备用。
具体的,所述卤代烃为含溴或碘的直链烷烃,卤代烃的碳原子数为3、5、6、8、10或12。如可以是溴丙烷、溴戊烷、溴代正己烷、溴代正辛烷、溴代正癸烷、溴代十二烷或碘丙烷等。
具体的,步骤1)中,所述吡唑与粉末状KOH、卤代烃的摩尔比为1∶1∶2。
本发明制备的一系列N,N-二烷基取代吡唑离子液体在室温下呈固态,熔点不超过120℃,具有良好的热稳定性,对空气稳定,对水稳定,几乎没有蒸气压,热分解温度在200℃左右,无黏度,可作为优良的催化剂催化CO2环加成反应。
利用上述N,N-二烷基取代吡唑离子液体催化合成碳酸丙烯酯的方法,其以环氧丙烷和CO2为原料,以N,N-二烷基取代吡唑离子液体为催化剂,通过CO2环加成反应合成碳酸丙烯酯;具体为:将N,N-二烷基取代吡唑离子液体与环氧丙烷按照1:50~600的摩尔比加入密闭高压反应釜中,通入CO2至反应釜内的压力为1~4MPa,然后在温度为90~130℃的条件下恒温恒压反应1~5h,反应结束经后处理即得碳酸丙烯酯。
进一步的,上述催化合成碳酸丙烯酯的方法,进一步优选如下:将N,N-二烷基取代吡唑离子液体与环氧丙烷按照1∶200的摩尔比加入密闭高压反应釜中,通入CO2至反应釜内的压力为2MPa,然后在温度为120℃的条件下恒温恒压反应4h,反应结束经后处理即得碳酸丙烯酯。
本发明设计合成了结构新颖的N,N-二烷基取代吡唑离子液体,并对其结构和理化性能进行了研究,然后利用这些离子液体作为催化剂催化CO2与环氧丙烷直接环加成合成环状碳酸酯的反应,并研究了它们的催化活性。试验结果发现:在较温和的条件下其具有良好的催化活性,未来在电化学、催化反应及作为分离萃取介质等方面具有广泛的应用前景。本发明对于吡唑离子液体的合成、性质及应用的研究具有重要的实用价值和学术意义。
附图说明
图1为本发明实施例1化合物N,N-二丙基吡唑溴盐的1H NMR图谱;
图2为本发明实施例1化合物N,N-二丙基吡唑溴盐的13C NMR图谱;
图3为本发明实施例2化合物N,N-二戊基吡唑溴盐的1H NMR图谱;
图4为本发明实施例6化合物N,N-二十二烷基吡唑溴盐的1H NMR图谱;
图5为本发明实施例7化合物N,N-二丙基吡唑碘盐的1H NMR图谱;
图6为本发明催化产物碳酸丙烯酯的1H NMR图谱;
另外:由于本发明所涉及的谱图太多,在此仅将代表性化合物的1H NMR图谱和13C NMR图谱列出。
具体实施方式
以下结合实施例对本发明的技术方案作进一步地详细介绍,但本发明的保护范围并不局限于此。
一,本发明所述N,N-二烷基取代吡唑离子液体的制备实施例列举如下:
其中,实施例1至7是关于吡唑溴盐离子液体的合成。
实施例1 N,N-二丙基吡唑溴盐的合成
取3.4g(0.05mol)的吡唑与2.8g(0.05mol)的粉末状KOH溶于乙腈中,在三颈瓶中常温搅拌1h后,通过恒压漏斗滴加12.3g(0.1mol)的溴丙烷,82℃反应24h。反应结束后,浓缩溶剂,CH2Cl2洗涤、过滤,滤液浓缩后乙酸乙酯洗涤、过滤,真空干燥得黄色固体N,N-二丙基吡唑溴盐。产率60%,熔点56.2℃。
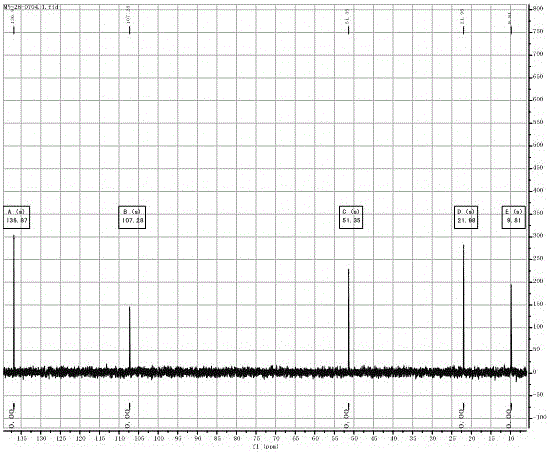
1H NMR(400 MHz, D2O) δ: 8.07 (d, 2H,Ar-H), 6.62 (t, 1H,Ar-H), 4.27 (t, 4H, NCH2CH2CH3), 1.79 (m, 4H, NCH2CH2CH3), 0.80 (t, 6H, NCH2CH2CH3)。13C NMR(101 MHz, D2O) δ:136.87, 107.28, 51.35, 21.98 , 9.81。MS(ESI):m/z 153.07 [C9H17N2+]; 81.80 [Br-]。
实施例2 N,N-二戊基吡唑溴盐的合成
取3.4g(0.05mol)的吡唑与2.8g(0.05mol)的粉末状KOH溶于乙腈中,在三颈瓶中常温搅拌1h后,通过恒压漏斗滴加15.1g(0.1mol)的溴戊烷,82℃反应24h。反应结束后,浓缩溶剂,CH2Cl2洗涤、过滤,滤液浓缩后乙酸乙酯洗涤、过滤,真空干燥得黄色固体N,N-二戊基吡唑溴盐。产率60%,熔点59.6℃。
1H NMR (400 MHz, D2O) δ: 8.07 (d, 2H), 6.63 (t, 1H), 4.31 (t, 4H), 1.77 (m, 4H), 1.19 (m, 8H), 0.76 (t , 6H)。MS(ESI):m/z 209.08 [C13H25N2+]; 81.80 [Br-]。
实施例3 N,N-二己基吡唑溴盐的合成
取3.4g(0.05mol)的吡唑与2.8g(0.05mol)的粉末状KOH溶于乙腈中,在三颈瓶中常温搅拌1h后,通过恒压漏斗滴加16.5g(0.1mol)的溴代正己烷,82℃反应24h。反应结束后,浓缩溶剂,CH2Cl2洗涤、过滤,滤液浓缩后乙酸乙酯洗涤、过滤,真空干燥得浅黄色固体N,N-二己基吡唑溴盐。产率60%,熔点30.2℃。
1H NMR (400 MHz, CDCl3) δ: 8.65 (d, 2H), 6.75 (t, 1H), 4.73 (t, 4H), 1.94 (m, 4H), 1.36 (m, 12H), 0.88 (t, 6H)。MS(ESI):m/z 237.14 [C15H29N2+]。
实施例4 N,N-二辛基吡唑溴盐的合成
取3.4g(0.05mol)的吡唑与2.8g(0.05mol)的粉末状KOH溶于乙腈中,在三颈瓶中常温搅拌1h后,通过恒压漏斗滴加19.3g(0.1mol)的溴代正辛烷,82℃反应24h。反应结束后,浓缩溶剂,CH2Cl2洗涤、过滤,滤液浓缩后乙酸乙酯洗涤、过滤,真空干燥得白色固体N,N-二辛基吡唑溴盐。产率40%,熔点112.4℃,热分解温度186.0℃。
1H NMR (400 MHz, DMSO-d6) δ: 8.60 (d, 2H), 6.93 (t, 1H), 4.47 (t, 4H), 1.82 (m, 4H), 1.27 (m, 20H), 0.80 ( m, 6H)。MS(ESI):m/z 293.26 [C19H37N2+]。
实施例5 N,N-二癸基吡唑溴盐的合成
取3.4g(0.05mol)的吡唑与2.8g(0.05mol)的粉末状KOH溶于乙腈中,在三颈瓶中常温搅拌1h后,通过恒压漏斗滴加22.1g(0.1mol)的溴代正癸烷,82℃反应24h。反应结束后,浓缩溶剂,CH2Cl2洗涤、过滤,滤液浓缩后乙酸乙酯洗涤、过滤,真空干燥得白色固体N,N-二癸基吡唑溴盐。产率40%,熔点115.5℃,热分解温度184.8℃。
1H NMR (400 MHz, DMSO-d6) δ: 8.57 (d, 2H), 6.93 (t, 1H), 4.45 (t, 4H), 1.80 (m, 4H), 1.24 (m, 28H), 0.85 (t, 6H)。MS(ESI):m/z 349.40 [C23H45N2+]。
实施例6 N,N-二十二烷基吡唑溴盐的合成
取3.4g(0.05mol)的吡唑与2.8g(0.05mol)的粉末状KOH溶于乙腈中,在三颈瓶中常温搅拌1h后,通过恒压漏斗滴加24.9g(0.1mol)的溴代十二烷,82℃反应24h。反应结束后,浓缩溶剂,CH2Cl2洗涤、过滤,滤液浓缩后乙酸乙酯洗涤、过滤,真空干燥得白色固体N,N-二十二烷基吡唑溴盐。产率40%,熔点117.2℃,热分解温度188.1℃。
1H NMR (400 MHz, CDCl3) δ: 8.58 (d, 2H), 6.74 (t, 1H), 4.73 (t, 4H), 1.93 (m, 4H), 1.47 (m, 4H), 1.25 (m, 32H), 0.88 (t, 6H)。MS(ESI):m/z 405.52 [C27H53N2+]。
实施例7 吡唑碘盐离子液体的合成(以N,N-二丙基吡唑碘盐的合成为例)
取3.4g(0.05mol)的吡唑与2.8g(0.05mol)的粉末状KOH溶于乙腈中,在三颈瓶中常温搅拌1h后,通过恒压漏斗滴加17g(0.1mol)的碘丙烷,82℃反应24h。反应结束后,浓缩溶剂,CH2Cl2洗涤、过滤,滤液浓缩后乙酸乙酯洗涤、过滤,真空干燥得黄色固体N,N-二丙基吡唑碘盐。产率60%,熔点83.7℃,热分解温度199.6℃。
1H NMR (400 MHz,CDCl3) δ: 8.54 (d, 2H), 6.78 (t, 1H), 4.81 (t, 4H), 2.03 (m, 4H), 1.09 (t, 6H)。13C NMR (101 MHz, D2O):δ 136.87 , 107.28 , 51.35 , 21.98 , 9.81。Anal. Calcd for C9H17N2I:C, 38.57; H, 6.07; N, 10.00. Found:C, 38.58; H, 6.32 ; N, 10.03. MS(ESI):m/z 153.07 [C9H17N2+]; 127.04 [I-]。
实施例8 吡唑四氟硼酸盐离子液体的合成(以N,N-二丙基吡唑四氟硼酸盐的合成为例)
单口瓶中加入4.66g(0.02mol) N,N-二丙基吡唑溴盐,20ml丙酮和2.64g(0.024mol)NaBF4,反应液变成淡黄色,回流搅拌6h,过滤,滤液浓缩,真空干燥得黄色固体N,N-二丙基吡唑四氟硼酸盐。产率88%。
MS(ESI): m/z 87.83 [BF4-]。
实施例9 吡唑六氟磷酸盐离子液体的的合成(以N,N-二丙基吡唑六氟磷酸盐的合成为例)
在250mL单口瓶中加入50mL蒸馏水和4.66g(0.02mol) N,N-二丙基吡唑溴盐,溶解后分批加入3.68g(0.02mol)KPF6,立即有细小的白色晶体生成,继续搅拌5h,抽滤,水洗(AgNO3检验,直至没有沉淀生成为止),干燥,得浅黄色固体N,N-二丙基吡唑六氟磷酸盐,产率67.7%。
MS(ESI): m/z 144.99 [PF6-]。
二,利用上述N,N-二烷基取代吡唑离子液体催化合成碳酸丙烯酯的方法。
实施例10
在100mL高压反应釜中加入10mL环氧丙烷,将N,N-二丙基吡唑溴盐按照与环氧丙烷1:200的摩尔比加入密闭高压反应釜中,密闭高压反应釜,通入CO2至釜内压力为2MPa,然后在120℃下恒温恒压反应4h。反应结束后反应釜冷却至室温,缓慢释放出CO2,反应产物经减压蒸馏除去未反应的环氧丙烷,过滤分离催化剂与产物,得到碳酸丙烯酯,收率82.3%。
1H NMR (400 MHz, CDCl3) δ: 4.89 (ddt, 1H), 4.59 (dd , 1H), 4.05 (dd, 1H), 1.50 (d, 3H)。
本实施例的反应方程式如下:
。
实施例11
参照实施例10,所用催化剂为N,N-二戊基吡唑溴盐,其他条件不变,得到的碳酸丙烯酯的收率为88.4%。
实施例12
参照实施例10,所用催化剂为N,N-二己基吡唑溴盐,其他条件不变,得到的碳酸丙烯酯的收率为77.6%。
实施例13
参照实施例10,所用催化剂为N,N-二辛基吡唑溴盐,其他条件不变,得到的碳酸丙烯酯的收率为68.6%。
实施例14
参照实施例10,所用催化剂为N,N-二癸基吡唑溴盐,其他条件不变,得到的碳酸丙烯酯的收率为73.4%。
实施例15
参照实施例10,所用催化剂为N,N-二十二烷基吡唑溴盐,其他条件不变,得到的碳酸丙烯酯的收率为64.0%。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种4‑氨基安替比林产品的合成工艺及其试液的配备方法与制造工艺
- 一种吡唑酮生产工艺的制造方法与工艺
- 一类含呋喃的苯基联吡唑甲酰胺类衍生物及其制备方法和用图
- N’?5?(取代苯基)?2?呋喃甲酰基?5?丙基?1h?吡唑?3?甲酸乙酯类化合物及其制备方法和应用
- 吡唑啉酮化合物及其用图
- 5-(4-氟苯基)-N–吗啉基-7-三氟甲基吡唑并[1,5-a]嘧啶-3-酰胺的合成方法
- 5-(4-氟苯基)-N–(4-氯苯基)-7-三氟甲基吡唑并[1,5-a]嘧啶-3-酰胺的合成方法
- 含醚性氧原子的全氟烷基取代吡唑环化合物及其制造方法
- 5-(4-氟苯基)-N–(4-氯苯乙基)-7-三氟甲基吡唑并[1,5-a]嘧啶-3-酰胺的合成方法
- 含有色酮结构的吡唑类n‐对硝基苯基马来酰亚胺衍生物及其制备方法与应用