血小板活化因子衍生物及其合成方法与流程
本发明属于化学合成技术领域,涉及一类血小板活化因子衍生物,具体涉及2-硫代脂肪链羧酸血小板活化因子及其衍生物的制备方法。
背景技术:
血小板活化因子(plateletactivityfactor,paf)的结构为醚键磷脂-1-o-烷基-2-乙酰基-sn-甘油-3-磷脂酰胆碱,如图1所示。paf分子量1100,其结构具有高度的立体特异性,且与其生物学效应密切相关,体内多种细胞可以合成。目前大多学者普遍认为,中性粒细胞、嗜碱性粒细胞、嗜酸性粒细胞、血小板、血管内皮细胞、肾髓质细胞、上皮细胞在激动剂(凝血酶、缓激肤、组胺、肿瘤坏死因子、白三烯、atp及血管紧张素i等)的作用下均能产生paf。paf作为一种特殊类型的细胞因子,能作用于多种组织和细胞,参与生殖、哮喘、过敏、炎症、肿瘤、休克等多种生理及病理效应,在体内发挥着类似于激素的生物学活性。
血小板活化因子于1972年由bemreniste等发现,是迄今为止一种最强活性的脂类介质。在研究初期,paf被认为与血小板的聚集和分泌有关,其诱导的血小板聚集过程不依赖于腺苷二磷酸(adp)或花生四烯酸(aa)的代谢产物txa2,被认为是诱导血小板聚集的第三条途径。但进一步的研究表明,paf特殊的受体与g蛋白耦联发生作用,paf受体还与细胞内信号传导途径有关,发挥着多种多样的生物功能。目前,paf在急性肝损害、肝纤维化和肝硬化的致病机制中的作用越来越受到重视,在众多参与肝脏疾病发病的炎性细胞因子中起着“中心放大”作用,并且通过诸多环节加重肝脏的损害。此外,paf可通过促进血栓形成,降低脑血流量,增加脑耗氧量,加重脑水肿,影响神经细胞功能并参与脑血管病的发生发展过程。
2-硫代血小板活化因子(2-s-paf)的化学名为1-氧-烷基-2-硫代乙酰基-sn-甘油-3-磷酸胆碱,是血小板活化因子paf的衍生物,如图1所示。2-s-paf可作为paf受体激动剂,其作用与pafc-18兔子血小板凝聚和pafc-16活化的豚鼠巨噬细胞相当。2-s-paf是溶血磷脂酶ii和paf-ah的底物,同时它也作为paf-ah检测试剂盒的底物。近年来,美国caymanchemical公司和日本azwell公司相继推出了检测血浆型paf-ah的试剂盒,由于其操作简单且无毒性,已逐步取代了传统的同位素定量检测法。
2-s-paf对paf的合成有着重要的作用,2-s-paf是以丙三醇为母体的一类化合物,其1位是烷基醚,2位是硫代乙酰基,3位是磷酸胆碱。这类化合物的化学结构类似于磷脂酰胆碱(卵磷脂),但又有不同之处,即卵磷脂的甘油分子中c1位连接的是饱和脂肪酸,c2位连接的是不饱和脂肪酸,而2-s-paf的甘油分子中c1位连接的是醚链脂肪醇,c2位连接的是硫代乙酰基。
masahirofuji等人以1,2-o-异亚丙基-sn-甘油为原料,并提供一个手性中心,经1位烷基化及丙酮叉的去保护得到二醇,二醇经3位的三苯甲基保护,2位与对硝基苯磺酰氯发生构型翻转,再经脱保护生成关键中间体paf-7,最后与磷酸胆碱反应得到最终产物。但此合成过程中,收率低,重排反应多。abulb.kazi等人认为重排是离去基活性不够,采取了比对硝基苯磺酰氯活性高的对甲磺酰氯发生构型翻转,优化了上述路线,以克服硫向氧的迁移问题。在paf-5的基础上,其认为最关键的是发生三苯甲基的迁移,将2位保护起来,再与磷酸胆碱反应,最后在催化剂的作用下,2位取代得到最终产物,但是,在对接开磷酰氯环时,收率不稳定,同时合成路线中用到了有毒的气体硫化氢,且中间产物极易氧化变质。masakazumurata等人以(s)-1-o-乙酰基-2-o-苄基甘油为原料,通过脂肪酶催化酯化,用作手性源,并选择四氢吡喃基保护伯羟基以防止环氧化物的形成和s→o酰基迁移,2位与对硝基苯磺酰氯发生构型翻转,再经ppts弱酸弱碱盐脱保护生成不稳定的关键中间体,最后与磷酸胆碱反应得到最终产物,但该合成路线需要保护去保护,综合收率低。
虽然现有研究已对2-s-paf做了些前瞻性的探索性工作,但现有合成路线仅适用于小规模制备,得到的2-s-paf量少,同时合成路线周期长,不能大批量合成,致使2-s-paf难以商品化,限制了2-s-paf的市场化应用。当前,2-s-paf的市场报价为3.5万元/100mg,这是由于其复杂的合成路线、难以量产造成的。因此,开发一种可以工厂化量产2-s-paf的合成路线,对于扩展2-s-paf的应用,将具有重要的现实意义,并且具有很大的市场潜力。
技术实现要素:
本发明针对2-s-paf现有合成路线周期长、难以大批量合成的技术缺陷,寻求一种高效、低成本、大批量合成高纯度2-s-paf的制备方法。本发明所述合成方法简便易于实施,合成路线扩张性强,同时可以快速获得2-硫代脂肪链羧酸血小板活化因子的多种衍生物。
本发明所述血小板活化因子衍生物,具有式(ⅰ)的化学结构:
式(ⅰ)中,取代基r代表酰基。
进一步地,所述式(ⅰ)中,取代基r代表脂肪族酰基。
作为取代基r的进一步优选,所述取代基r代表链状脂肪族酰基,r=cnh2n+1co-,n为正整数且表示碳链的长度。
为了详实的描述式(ⅰ)血小板活化因子衍生物的结构,本发明对上下文中的术语进行解释说明,但该解释并不代表对本发明保护范围的特别限定。
术语“酰基”应理解为有机或无机含氧酸分子中去掉羟基后,剩下的一价原子团的统称,或是理解为羧酸脱去羟基后的剩余部分。通常的酰基化学式为r′-c=o-。酰基具有较强的化学反应活性,可与卤素原子、烷氧基、氨基或取代氨基及酰氧基结合,分别获得酰卤、酯、酰胺和酸酐。乙酰基为一种常见的酰基,化学式为ch3-c=o-或记为ac-,是一个由甲基和羰基组成的酰基官能团。
术语“脂肪族酰基”应理解为酰基r′为脂肪族取代基。脂肪族取代基是由脂肪族化合物形成的取代基。脂肪族化合物是链状烃类(开链烃类)及除芳香族化合物以外的环状烃类及其衍生物的总称。其中,属于脂肪族的碳环化合物又称脂环族化合物。
术语“链状脂肪族酰基”应理解为酰基r′为链状脂肪族取代基。链状脂肪族化合物可以理解为链状烃类(开链烃类)及其衍生物的总称。链状脂肪族化合物包括饱和的链状烃类,比如烷烃类化合物,还包括含有不饱和键的链状烃类。这里所述的不饱和键具有广义的概念,比如可以是碳碳双键、碳氧双键、碳碳叁键等。取代基r=cnh2n+1co-,n为正整数且表示碳链的长度,说明链状脂肪族酰基是指饱和的链状烃类形成的酰基,比如上述的乙酰基,就是一种简单的链状脂肪族酰基,其n=1。
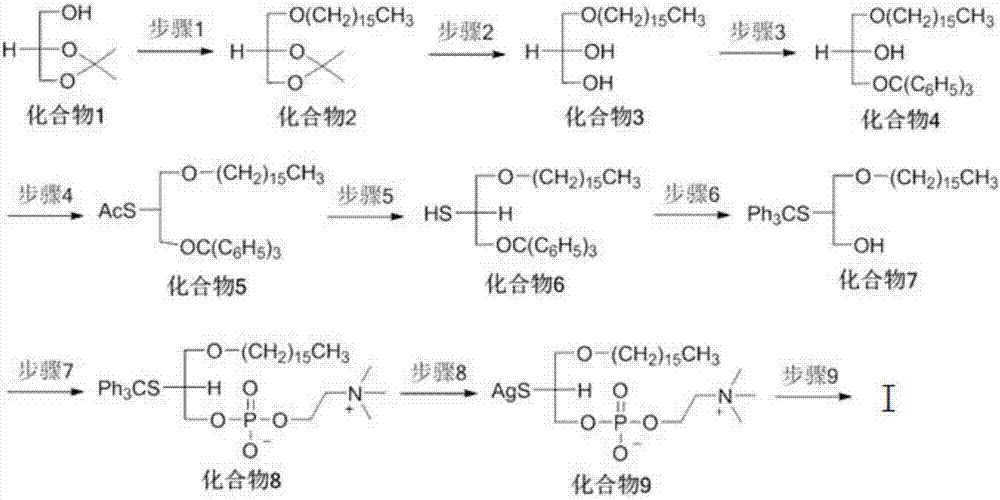
另一方面,本发明还给出了所述血小板活化因子衍生物的制备方法。式(ⅰ)化合物的路线如图2所示。为了叙述方便,本发明的中间产物均按照化合物1,2,3…进行编号。
上述合成路线中,步骤1,以化合物1(s)-丙酮缩甘油为反应起始化合物,向溶有化合物1的溶剂中,加入十六烷衍生物,在碱试剂存在下进行反应,得到化合物2。在本步骤中,化合物1的反应溶剂可以选用多种,优选地,可以溶解于二甲基甲酰胺。十六烷衍生物主要是指卤代十六烷,优选为1-溴代十六烷或者1-碘代十六烷,或是二者的混合物;也可以选用十六烷基磺酸酯。本步反应在碱性条件下进行,可以选用的碱试剂为nah、kh、koh、naoh、naot-bu中的一种或是其混合碱,优选为nah。
上述合成路线中,步骤2,化合物2酸性水解,得到化合物3。化合物2的化学名为(r)-丙酮缩甘油十六烷醚。化合物2进行酸性水解所需的酸为盐酸、醋酸、硫酸、对甲苯磺酸、樟脑磺酸中的一种或是其混合酸。优选地,化合物2进行酸性水解所需的酸选用浓度为0.5m的盐酸。
上述合成路线中,步骤3,选择性的保护化合物3的末端羟基,得到化合物4。化合物3的化学名为(r)-3-(十六烷氧基)丙烷-1,2-二醇。本步骤需要特定的反应试剂和催化剂。步骤3的反应溶剂为mecn/四氢呋喃(v/v=1/5),所用催化剂为三乙胺、吡啶、4-二甲胺基-吡啶、2,6-二甲基吡啶中的一种或是其混合物,所使用的末端羟基保护试剂为三苯基氯甲烷、三苯基溴甲烷中的一种或是其混合物。
上述合成路线中,步骤4,将化合物4在溶剂中生成离去基,向中间体中加入硫代试剂,得到化合物5。化合物4的化学名为(s)-1-(十六烷氧基)-3-三苯甲氧基丙烷-2-醇。本步骤的催化剂为三乙胺,溶剂为二氯甲烷,生成离去基的试剂为甲烷磺酰氯、对甲苯磺酰氯、对硝基苯磺酰氯、邻硝基苯磺酰氯、2,4-二硝基苯磺酰氯苯中的一种或是其混合物;所述中间体的反应溶剂为二甲基甲酰胺,所使用的硫代试剂为硫代乙酸盐。这里的硫代乙酸盐为硫代乙酸钾、硫代乙酸钠、硫代乙酸锂、硫代乙酸三乙胺盐中的一种或是其混合物。
上述合成路线中,步骤5,化合物5在碱存在下脱去乙酰基,得到化合物6。化合物5的化学名为(r)-s-(1-(十六烷氧基)-3-(三苯甲氧基)丙烷-2硫醇乙酸酯。本步骤的反应溶剂为甲醇/四氢呋喃(v/v=2/1),所用碱试剂为28%甲醇钠的甲醇溶液,或是固体甲醇钠、固体乙醇钠,或者定量的氢氧化锂/过氧化氢体系。
上述合成路线中,步骤6,化合物6在二氯甲烷溶液中,加入反应试剂bf3et2o,进行三苯甲基迁移反应,得到化合物7。化合物6的化学名为(r)-s-(1-(十六烷氧基)-3-(三苯甲氧基)丙烷-2硫醇。本步骤所用溶剂是苯、甲苯、二甲苯、环己烷、二氯甲烷、二氯乙烷、1,1,2,2-四氯乙烷中的一种或是其混合物,所使用的催化剂为三氯化铝、三氟化硼、三氟化硼乙醚,二氯化锌中的一种或是其混合物。
上述合成路线中,步骤7,在溶有化合物7的溶剂中,加入催化剂,反应完全,得到化合物8。化合物7的化学名为(s)-s-(3-(十六烷氧基)-2-(三苯甲氧基)丙烷-1醇。本步骤所用反应溶剂为苯、甲苯、二甲苯、环己烷中的一种或是其混合物,所述催化剂为三乙胺。
上述合成路线中,步骤8,向化合物8的反应溶剂中,加入掉保护基催化剂,得到化合物9。化合物8的化学名为(r)-3-(1-(十六烷氧基)-2-(三苯甲硫基)丙基-2-(三甲胺基乙基)膦酸酯。本步骤所用反应溶剂为四氢呋喃、乙腈、吡啶中的一种或是其混合物,所述掉保护基催化剂为硝酸银、乙酸银、三氟乙酸银中的一种或是其混合物。
上述合成路线中,步骤9,化合物9与酰化试剂反应,得到目标产物ⅰ。化合物9的化学名为(r)-(1-(十六烷氧基)-3-(氧代(2-(三甲胺基乙氧基)膦酸氧基丙基)-1-硫化银。本步骤所用反应溶剂为二氯甲烷、四氢呋喃、乙腈、吡啶中的一种或是其混合物,所用催化剂为碘化钾、4-二甲胺基-吡啶(dmap)中的一种或是其混合物,所述酰化试剂为酸酐或是酰氯。在本步骤中,目标产物ⅰ的结构取决于选用的酰化试剂,通过选择不同的酰化试剂,即可得到2-s-paf的衍生物。
上述血小板活化因子衍生物的合成路线也是在合成此类化合物中首次成功应用,该合成路线新颖、产率高并且产物易于分离,可以说是制备此类化合物的最优路线。式(ⅰ)结构的血小板活化因子衍生物的具体合成方法,在实施例中有详细的描述。
与现有技术相比,本发明所述血小板活化因子衍生物及其合成方法具有下述的有益效果或优点:
(1)本发明采取价格便宜的手性源s构型的丙酮缩甘油作为起始反应物,经过9步反应,以8.11%的总收率合成了2-s-paf,有效地解决了其原有合成路线长、产率低、原料价格昂贵的技术缺陷。
(2)采用本发明的合成路线,选用不同的酰化试剂,可以快速合成其它多种血小板活化因子衍生物。
(3)2-s-paf及其衍生物具有高度的立体特异性,可作用于多种组织和细胞,参与生殖、哮喘、过敏、炎症、肿瘤、休克等多种生理及病理效应,在体内发挥着类似于激素的生物学活性。但其合成困难、来源缺乏等因素限制了其活性研究以及临床应用。本发明优化了其合成方案,实现低成本、大批量合成2-s-paf及其衍生物。这对于2-s-paf及其衍生物的活性研究、生物实验、临床应用和疾病治疗具有重大意义。
附图说明
图1是本发明所述血小板活化因子的分子结构图。
图2是本发明所述血小板活化因子衍生物的制备路线图。
图3是本发明所述2-s-paf衍生物的合成路线图。
在以下实施例中进一步描述本发明,而不以任何形式旨在限制如权利要求所表明的本发明的保护范围。
具体实施方式
实施例1,2-s-paf的合成
本实施例给出2-s-paf的合成路线图,具体见图3。最终产物2-s-paf用tm表示。
本实施例所给合成路线中,步骤1的具体操作为,在圆底烧瓶中加入溶解有nah(7.26g,0.182mol)的二甲基甲酰胺(dmf)溶液300ml,再加入化合物1(20.0g,0.151mol),搅拌30min;再向反应液中加入十六烷基磺酸酯(58.21g,0.182mol),室温反应12小时;处理反应液,萃取得到有机相,硅胶柱纯化(洗脱剂为正己烷/乙酸乙酯,v/v=95/5),得到39.0g化合物2,产率85%。
1hnmr(cdcl3)δ0.88(t,j=6.4hz,6h),1.30(m,28h),1.56(m,5h),3.63(m,5h).
步骤2的具体操作为,将化合物2(52.37g,0.147mol)溶于370ml的丙酮中,加入150ml的盐酸(0.5m);混合液回流搅拌2h,待反应完全后,改为蒸馏装置,蒸馏出大部分丙酮,然后析出大量固体;此固体用水、饱和碳酸氢纳溶液、水、乙醚清洗,干燥得到44.37g白色固体化合物3,产率95%。
1hnmr(cdcl3)δ0.93(t,j=6.4hz,3h),1.29(m,26h),1.62(m,2h),1.80-2.43(2h),3.50-3.92(m,7h).
步骤3的具体操作为,将21.5g化合物3(67.9mmol)溶于250mlthf和50mlmecn中,再加入22.5g三苯基溴甲烷、20mlet3n,回流反应15h;反应完全后,将反应液浓缩,过滤除掉三乙胺的盐酸盐,并用少量干燥的乙酸乙酯洗涤沉淀;滤液分别用h2o、1%hcl、饱和nahco3溶液、饱和食盐水洗涤;无水硫酸镁干燥,旋干,过柱(pe/ea=10/1~5/1),加入石油醚,放置于冰箱过夜,有白色固体析出,过滤得到43.6g化合物4,产率78%。
1hnmr(cdcl3)δ0.94(t,j=6.8hz,3h),1.23-1.44(m,26h),1.40-1.65(m,2h),2.43(d,j=4.8hz,1h),3.10-3.25(m,2h),3.35-3.60(m,4h),3.88-4.03(m,1h),7.17-7.55(m,15h).
步骤4的具体操作为,在-5℃下,将26.2g(46.8mmol)化合物4溶于270mldcm中,并同时逐滴加入9.8mlet3n和3.6ml甲磺酰氯,仔细控制同时加完,然后在此温度反应2h;反应混合物被倒入冰水中,分离有机相,用二氯甲烷萃取无机相,合并有机相;分别用0.1mhcl、饱和nahco3溶液、饱和nacl溶液洗涤;无水mgso4干燥,浓缩得到中间产物。将28.7g中间产物溶于240mldmf中,加入7.6ghsac和3mlet3n进行反应。水浴温度设定在90℃,12h后,再补加3.8ghsac和1.5mlet3n继续反应4h后,反应完全。冷却,并将反应液倒入冰水中,并用乙酸乙酯萃取,有机相分别用水和饱和nacl溶液洗涤,无水mgso4干燥,旋干,过柱(pe/ea=100/1~20/1),得到17.1g化合物5,产率57%。
1hnmr(cdcl3)δ0.88(t,j=6.4hz,3h),1.10-1.40(m,26h),1.40-1.60(m,2h),2.31(s,3h),3.22(dd,j=9.2,5.6hz,1h),3.37(t,j=6.6hz,2h),3.39(dd,j=9.2,3.8hz,1h),3.65(d,j=6.0hz,2h),3.80-3.97(m,1h),7.15-7.50(m,15h).
步骤5的具体操作为,将13.4g化合物5(21mmol)溶于78mlmeoh和39mlthf中,并于0℃下逐滴加入4.6ml质量分数为28%的meona溶液。30min后,反应完全,浓缩,并用acoet稀释浓缩液,溶液分别用h2o、0.1mhcl、饱和nahco3溶液、饱和nacl溶液洗涤。无水mgso4干燥,旋干,过柱(pe/dcm=50/1~20/1),得到9.6g化合物6,产率77%。
1hnmr(cdcl3)δ0.88(t,j=6.4hz,3h),1.10-1.40(m,26h),1.40-1.65(m,2h),1.89(d,j=8.2hz,sh),2.95-3.15(m,1h),3.26(dd,j=9.2,5.8hz,1h),3.32(dd,j=9.2,5.4hz,1h),3.40(t,j=6.6hz,2h),3.55-3.70(m,2h),7.15-7.50(m,15h).
13cnmr(cdcl3)δ14.12,22.70,26.16,29.38,29.54,29.62,29.65,29.68,29.72,31.95,39.88,64.86,71.34,72.57,86.63,127.07,127.84,128.77,144.06.
步骤6的具体操作为,将9.6g化合物6(16.7mmol)溶于180mldcm中,在-10℃下,向反应体系中逐滴加入2.4mlbf3.oet2。45min后,反应混合物倒入冰饱和nahco3溶液中,dcm萃取,有机相用饱和nacl洗涤,无水mgso4干燥,旋干,过柱(pe/ea=8/1~4/1),得到7.1g化合物7,产率74%。
1hnmr(cdcl3)δ0.88(t,j=6.4hz,3h),1.10-1.35(m,26h),1.35-1.55(m,2h),2.54-2.72(m,2h),3.00-3.58(m,6h),7.15-7.35(m,9h),7.40-7.50(m,6h).
13cnmr(cdcl3)δ14.13,22.71,26.05,29.39,29.44,29.52,29.59,29.64,29.69,29.72,31.95,45.42,64.92,67.46,71.47,72.94,126.80,128.00,129.58,144.88.
步骤7的具体操作为,将化合物7(9.98g,17.33mmol)和三乙胺(5.5ml,39.45mmol)溶于苯中,再将100ml的2-氯-2-氧-1,3,2-二氧磷杂环戊烷(4.69g,32.88mmol)的苯溶液加入到化合物7的溶液中,室温搅拌反应3.5h,过滤除去沉积盐,滤液浓缩得到中间产物,将中间产物溶于24ml浓度为3.8m三甲胺的乙腈溶液中,50℃反应16h。反应液浓缩,硅胶柱纯化。洗脱剂为氯仿/甲醇/水(v/v/v=32/9/1~20/15/1),得8.35g化合物8,产率65%。
1hnmr(dmso-d6)δ0.89(t,j=6.4hz,3h),1.10-1.50(m,28h),2.50-2.65(m,1h),2.84(dd,j=10.2,4.4hz,1h),3.05-3.27(m,3h),3.18(s,9h),3.50-3.65(m,2h),3.75-3.95(m,2h),4.10-4.30(m,2h),7.15-7.40(m,9h),7.45-7.55(m,6h).
13cnmr(dmso-d6)δ14.15,22.70,26.07,29.38,29.56,29.68,29.74,31.93,44.93,54.29,59.18,64.87,66.22,67.20,69.35,70.80,126.70,127.94,129.65,144.88.
步骤8的具体操作为,将化合物8(1.2g,7.62mmol)溶于12ml的mecn和1.2mlmeoh中,再加入0.3ml吡啶,搅拌。将agno3(0.55g,3.24mmol)的mecn(1.8ml)溶液加入到反应液中,反应1h。向反应液中加入乙醚,抽滤,用乙腈/乙醚(v/v=1/1)清洗,真空干燥,得到0.6g化合物9,产率61%。
步骤9的具体操作为,将化合物9(115mg,0.19mmol)溶于2ml的mecn中,加入ki(38mg,0.38mmol),dmap(46mg,0.38mmol)和乙酸酐(39mg,0.38mmol),反应16h得到产物。用chcl3/meoh作为洗脱剂,柱层析分离纯化,得到目标产物2-s-paf,产率61%。
1hnmr(cdcl3)δ0.88(t,j=6.4hz,3h),0.89(t,j=6.4hz,3h),1.20-1.42(m,32h),1.42-1.60(m,2h),1.60-1.80(m,2h),1.96-2.18(m,4h),2.54(t,j=7.6hz,2h),2.72-2.92(m,6h),3.30-3.47(m,2h),3.40(s,9h),3.48-3.68(m,2h),3.75-4.03(m,5h),4.26-4.42(m,2h),5.24-5.50(m,8h).
13cnmr(cdcl3)δ14.09,14.13,22.59,22.71,25.46,25.66,26.11,26.43,27.24,29.33,29.38,29.58,29.70,29.74,31.53,31.94,43.63,44.20,54.51,59.24,64.24,66.52,69.20,71.47,127.53,127.83,128.07,128.34,128.65,128.68,129.10,130.52,198.98.
实施例2,一种2-s-paf衍生物的合成
本实施例所给合成路线参见图3,步骤1~8的具体操作参见实施例1。
步骤9的具体操作为,将化合物9(30mg,0.05mmol)溶于2ml的mecn中,加入ki(8.3mg,0.05mmol),dmap(6mg,0.05mmol)和十六碳酰氯(137mg,0.5mmol),反应16~24h得到产物。用chcl3/meoh作为洗脱剂,柱层析分离纯化,得到2-s-paf衍生物,产率61%。
1hnmr(cdcl3)δ0.88(t,j=6.4hz,3h),0.89(t,j=6.4hz,3h),0.91(t,j=6.4hz,3h),1.20-1.42(m,64h),1.42-1.60(m,2h),1.60-1.80(m,2h),1.96-2.18(m,4h),2.33(t,j=7.6hz,2h),2.54(t,j=7.6hz,2h),2.72-2.92(m,6h),3.30-3.47(m,2h),3.40(s,9h),3.48-3.68(m,2h),3.75-4.03(m,5h),4.26-4.42(m,2h),5.24-5.50(m,8h).
上面结合实施例对本发明做了进一步的叙述,但本发明并不限于上述实施方式,在本领域的普通技术人员所具备的知识范围内,还可以在不脱离本发明宗旨的前提下做出各种变化。
- 还没有人留言评论。精彩留言会获得点赞!