一种右旋兰索拉唑晶型及制备方法与流程
本发明涉及一种右旋兰索拉唑晶型及制备方法。
背景技术:
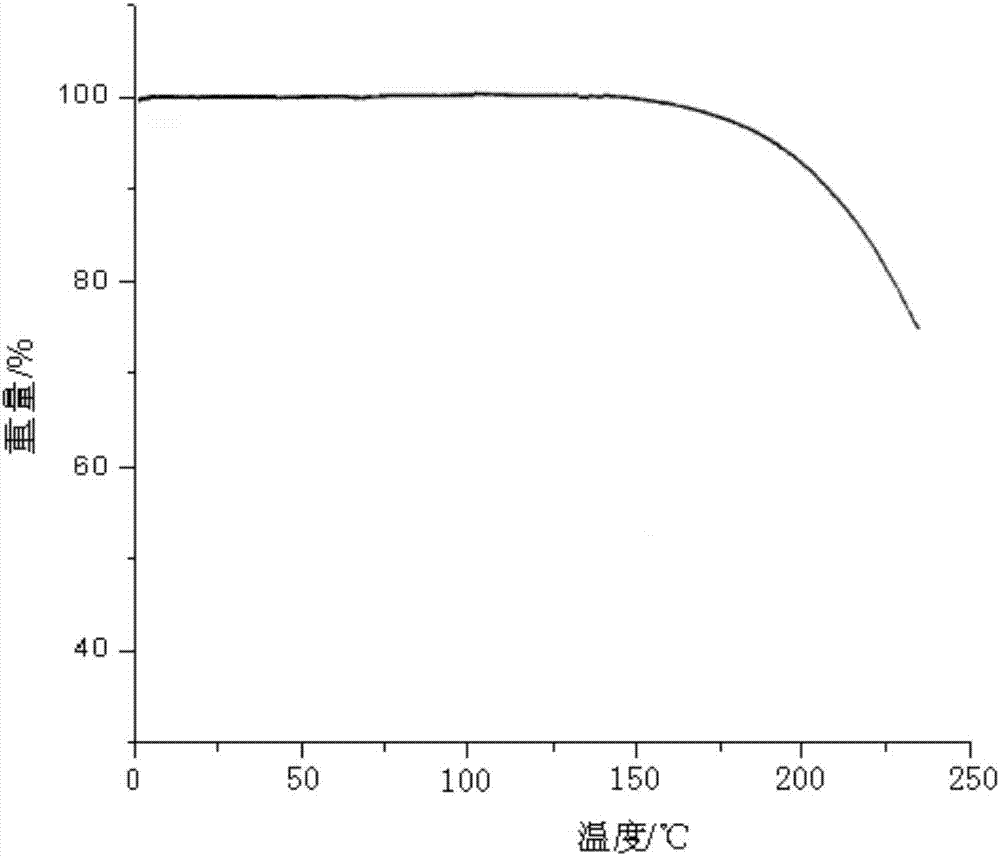
:右旋兰索拉唑是一种新型的质子泵抑制剂,是兰索拉唑的右旋对映体,由日本武田制药公司开发,用于治疗与非糜烂性胃食管反流病相关的胃灼热及不同程度的糜烂性食道炎。化学结构如下:质子泵抑制剂为苯并咪唑类衍生物,特异性和非竞争性的作用于H+/K+-ATP酶,治疗消化性溃疡。质子泵抑制剂多为脂溶性弱碱性,吸收入血后进入壁细胞分泌小管、小管泡腔中酸性环境后,活化产物一般为活性次磺酸和次磺酰胺,与H+-K+-ATP酶巯基偶联形成一个不可逆的共价二硫键,阻断H+-K+转运机制,从而抑制胃酸分泌。专利CN102875531B公开了右旋兰索拉唑原料在乙酸乙酯、正庚烷、二乙胺体系中加热溶解然后降温析晶得到的晶型产品,但本申请人采用此专利方法制得的晶型稳定性在工业应用上存在着产品不稳定的问题,因此努力寻求稳定的右旋兰索拉唑晶型是有必要的。本发明人通过购买市售右旋兰索拉唑原料进行大量的试验,发现本发明采用的制备方法获得的晶型,与外购原料和专利CN102875531B所述制备的样品相比,本发明制备的结晶表现出异乎寻常的稳定性,且溶解度及溶出度也得到极大的改善。技术实现要素:本发明目的是提供一种右旋兰索拉唑晶型及制备方法,所制得的产品稳定性、溶解度及溶出度高于现有技术,适合工业化生产。本发明提供一种右旋兰索拉唑晶型,使用Cu-Kα辐射,以2θ角度表示的X-射线粉末衍射在2.7±0.2°、10.8±0.2°、14.3±0.2°、21.2±0.2°、22.9±0.2°、27.8±0.2°处有特征峰。进一步地,右旋兰索拉唑晶型具有如图1所示的X-射线粉末衍射谱图。又进一步,一种右旋兰索拉唑的晶型,其制备方法包括如下步骤:1)将无水右旋兰索拉唑粗品溶于10倍(ml/g)的丙酮中,搅拌溶解;2)加入0.05倍(g/g)的活性炭,搅拌脱色0.5小时,过滤;3)取上述步骤2)的滤液在一定温度下加入第一部分异丙醚,搅拌培养晶核,逐渐有微小晶粒出现,然后滴加第二部分异丙醚,滴加时间3小时,搅拌析晶完全;4)过滤,真空干燥得到白色右旋兰索拉唑晶体。更进一步地,步骤3)中温度为20~30℃;培养晶核的时间0.5~1小时,优选0.5小时。第一部分异丙醚的量与右旋兰索拉唑粗品的体积质量比为4~7:1ml/g,优选6:1ml/g;第二部分异丙醚与右旋兰索拉唑粗品的体积质量比为30~35:1ml/g。本发明与现有技术相比,具有如下优点:1)本发明所采用丙酮、异丙醚为低毒的第三或第四类溶剂,溶剂毒性小,药物安全性高,不会出现结晶体系粘稠现象,成本低廉,有利于工业放大和药物生产。2)本发明采用先培养晶核再缓慢析晶方法,获得了稳定的晶体,分段加入异丙醚方式避免由于析晶速度快将杂质包含产品中,又能得到重现性好的晶型。3)本发明对右旋兰索拉唑进行了加速实验、高温影响因素试验研究,其含量未见明显降低,最大单杂未见明显增加,各项指标均无明显变化,说明本品具有良好的稳定性,重现性,便于长期储存,临床用药更为安全。4)本发明人还发现右旋兰索拉唑相比现有的晶型水溶性及溶出度得到改善,有利于临床的使用。附图说明图1是实施例1的右旋兰索拉唑晶型X-射线粉末衍射图谱。图2是实施例1的右旋兰索拉唑晶型热重分析图谱。具体实施方式以下实施例仅用于进一步说明本发明,但不限制本发明。实施例1右旋兰索拉唑晶型的制备向1000ml烧瓶中加入10g右旋兰索拉唑粗品,100ml丙酮,搅拌溶解。加入0.5g活性炭,搅拌脱色0.5小时,过滤。控制温度在20℃条件下,向滤液加入40ml异丙醚,搅拌养晶0.5小时,有微小晶粒出现,溶液变浑浊。继续滴加300ml异丙醚,3小时滴加完毕。过滤,真空干燥得白色结晶性粉末8.99g,HPLC纯度99.91%。所制得的右旋兰索拉唑用粉末X射线衍射测定法测定,得到X-射线粉末衍射图谱如图1所示。采用美国Perkin-Elmer公司PEPyrisDiamondTG热重分析仪,热重分析图谱如图2所示,晶型熔点为146-153℃,右旋兰索拉唑晶型在降解之前无重量损失,可以推断为右旋兰索拉唑的无水合物。实施例2右旋兰索拉唑晶型的制备向1000ml烧瓶中加入10g右旋兰索拉唑粗品,100ml丙酮,搅拌溶解。加入0.5g活性炭,搅拌脱色0.5小时,过滤。控制温度在20℃条件下,向滤液加入60ml异丙醚,搅拌养晶1小时,有微小晶粒出现,溶液变浑浊。继续滴加350ml异丙醚,3小时滴加完毕。过滤,真空干燥得白色结晶性粉末9.21g,HPLC纯度99.97%。根据XPRD数据,所得晶型与实施例1中晶型一致。采用美国Perkin-Elmer公司PEPyrisDiamondTG热重分析仪得到的热重分析图谱与实施例1一致。实施例3右旋兰索拉唑晶型的制备向1000ml烧瓶中加入10g右旋兰索拉唑粗品,100ml丙酮,搅拌溶解。加入0.5g活性炭,搅拌脱色0.5小时,过滤。控制温度在20℃条件下,向滤液加入70ml异丙醚,搅拌养晶0.5小时,有微小晶粒出现,溶液变浑浊。继续滴加300ml异丙醚,3小时滴加完毕。过滤,真空干燥得白色结晶性粉末9.08g,HPLC纯度99.93%。根据XPRD数据,所得晶型与实施例1中晶型一致。采用美国Perkin-Elmer公司PEPyrisDiamondTG热重分析仪得到的热重分析图谱与实施例1一致。实施例4右旋兰索拉唑晶型的制备向1000ml烧瓶中加入10g右旋兰索拉唑粗品,100ml丙酮,搅拌溶解。加入0.5g活性炭,搅拌脱色0.5小时,过滤。控制温度在30℃条件下,向滤液加入50ml异丙醚,搅拌养晶1小时,有微小晶粒出现,溶液变浑浊。继续滴加350ml异丙醚,3小时滴加完毕。过滤,真空干燥得白色结晶性粉末7.56g,HPLC纯度99.86%。根据XPRD数据,所得晶型与实施例1中晶型一致。采用美国Perkin-Elmer公司PEPyrisDiamondTG热重分析仪得到的热重分析图谱与实施例1一致。试验例1:稳定性试验1)加速试验本实验按照中国药典2015版第二部附录ⅪⅩC原料药与药物制剂稳定性试验指导原则进行,加速试验条件:温度40℃±2℃,相对湿度75%±5%。试验结果见表1。表1:加速试验结果对比其中,样品1是实施例1产品;样品2是实施例2产品;样品3是参照专利CN102875531B方法制得的右旋兰索拉唑晶型;样品4是市售右旋兰索拉唑原料药,产自上海汇伦生命科技有限公司。由表1加速稳定性试验表明,本发明右旋兰索拉唑的新晶型40℃加速试验结果表明,本发明所制得晶型与现有技术方法制得晶型相比,各项检测指标均无显著性变化,性质稳定。2)高温影响因素试验分别取1g右旋兰索拉唑样品1、样品2、样品3和样品4,置于洁净容器中,在温度60℃,相对湿度75%的条件下放置20天,分别在0、5、10、20天取样,测定右旋兰索拉唑纯度及杂质情况。表2:60℃影响因素试验结果其中,样品1是实施例1产品;样品2是实施例2产品;样品3是参照专利CN102875531B方法制得的右旋兰索拉唑晶型;样品4是市售右旋兰索拉唑原料药,产自上海汇伦生命科技有限公司。由表2结果表明,右旋兰索拉唑在高温条件下放置20天后,本发明所制得的右旋兰索拉唑晶型与其它传统的右旋兰索拉唑晶型相比,其杂质的含量明显低于其它右旋兰索拉唑晶型,稳定性较高。试验例2:溶解度试验按照中国药典2015年版二部“凡例”规定的方法做了以下几种溶剂的溶解度试验。方法:称取研成细粉的各样品适量(精确至±2.0%),加入一定量体积的水,在25±2℃室温下每隔5分钟振摇30秒,观察30分钟内溶解情况,结果见表3。表3:溶解度试验结果样品编号溶解度(mg/ml)样品12.39样品22.66样品30.99样品40.67其中,样品1是实施例1产品;样品2是实施例2产品;样品3是参照专利CN102875531B方法制得的右旋兰索拉唑晶型;样品4是市售右旋兰索拉唑原料药,产自上海汇伦生命科技有限公司。从试验结果可以看出,本发明与现有技术相比,本发明制得的右旋兰索拉唑晶型在水中的溶解性增大。试验例3:溶出度考察溶出度测定方法:将样品1、样品2、样品3和样品4按照《中华人民共和国药典》2015年版二部附录XC法第二法考察溶出度。以pH6.8磷酸盐缓冲液900mL为溶出介质,温度为37℃,转速为75r·min-1,每个待测样品中的药物量为30mg。采用紫外分光光度法分别于5、10、15、20、30、45、60min取样进行溶出度测定,检测波长为286nm,在5~25mg·L-1内线性关系良好,回收率、精密度实验均符合方法学要求。结果见表4。表4:右旋兰索拉唑的溶出度考察结果其中,样品1是实施例1产品;样品2是实施例2产品;样品3是参照专利CN102875531B方法制得的右旋兰索拉唑晶型;样品4是市售右旋兰索拉唑原料药,产自上海汇伦生命科技有限公司。从表4试验结果可以看出,与现有技术的右旋兰索拉唑相比,本发明所提供的右旋兰索拉唑晶型溶出度得到显著改善。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1