一种从土壤中特异性分离酸杆菌的方法与流程
本发明涉及的领域为现代分子生物学技术和微生物技术的交叉领域,具体的说为一种基于特异性引物-pcr结合改良后的基本培养基平板特异性分离酸杆菌的方法。
背景技术:
酸杆菌(acidobacteria)最初的发现是由日本的kishimoto等于1991年发现,酸杆菌作为土壤微生物群落中的重要组成部分,其相对丰度仅次于土壤中的变形菌门,约占土壤细菌总量的20~50%,而在某些特定环境中,如栗树根际土壤中,作为土壤中的绝对优势微生物类群,其相对丰度可达到65%。酸杆菌门在环境的分布极为广泛,包括自然环境以至于一些极端环境与污染环境,而且酸杆菌在驱动土壤物质循环和生态环境构建过程当中具有重要的生态学意义。
近几年的研究发现根据系统发育研究结果可以将酸杆菌下分为27种不同纲水平微生物。对土壤微生物宏基因组测序表明,酸杆菌具有降解植物中纤维素相关基因,以此推测在环境较为恶劣的情况下,如酸性、低温环境中,相比于其他微生物,对恶劣环境强适应性的酸杆菌所具备的纤维素降解能力就尤为突出;酸杆菌对于所处生境中的铁元素循环也起到积极作用,如可在葡萄糖发酵过程中形成fe(ⅱ),为发酵过程中的其他微生物提供必要的养分。
酸杆菌的纯化菌株很难通过人工培养获得,因此对于该类群微生物的表型、生理特征以及机理研究极为稀少。近年来针对酸杆菌的研究多是以分子生物学手段为基础,通过16srdna对酸杆菌的遗传多样性开展了诸多研究。但是关于通过培养实验以快速获得酸杆菌单菌株的方法尚未见报道,极大的影响了对于酸杆菌的生态学价值的研究,并降低了探索其潜在其他价值的可能性。
目前,国内外以16srdna为基础对酸杆菌开展的研究多是以细菌的通用引物为主,而细菌的通用引物虽然可以在门水平上为微生物多样性的划分提供依据,但是在更为精细的分类水平上,则需要设计针对该门类微生物的特异性引物。因此目前仍然缺乏具备足够特异性的高效引物完成对酸杆菌16srdna的特异性扩增。
技术实现要素:
本发明旨在提供一种基于特异性引物-pcr结合改良后的mm平板特异性分离酸杆菌的方法。
为完成以上目标,本发明的基本方案为:
一种基于门水平的特异性引物-pcr结合改良后的基本培养基平板特异性分离酸杆菌的方法。方法为,将样品悬浊液稀释涂布到改良后的固体基本培养基平板,选择白色单菌落继续采用连续划线平板法纯化,将纯化后的单菌落经液体培养基扩大培养,提取基因组dna,应用酸杆菌门特异性引物pcr扩增16srdna片段,若得到片段大小为1462的阳性扩增产物,即表明筛选得到该样品中的酸杆菌。
酸杆菌门特异性引物acido_31f:5‘-gatcctggctcagaatc-3’,acido_1492r:5‘-ggttaccttgttacgactt-3’
具体操作方案为:
1)土壤悬浊液的制备:首先称取5g待测土壤至于50ml已灭菌的锥形瓶并添加少量玻璃珠,而后向其中加入45ml生理盐水于150rpm条件下振荡90min,使土壤微生物充分溶于生理盐水中;振荡结束后,于室温下静置30min,采用生理盐水对静置后的土壤上清液按照10倍的梯度进行连续稀释;
2)涂布稀释后的上清液至改良后的mm固体培养基:吸取上一步中得到的稀释后的样液(稀释度以10-4为宜),采用涂布平板法涂布,于30℃下培养3~6天;
3)连续划线平板法纯化:接种环挑取上一步中得到的单菌落,在改良后的固体基本培养基上连续划线以达到分离纯化的目的;
4)纯培养菌株基因组dna的提取:将步骤3中分离纯化后得到的单菌落接种于改良后的液体基本培养基,30℃下150rpm振荡培养过夜,而后采用溶菌酶、蛋白酶k和氯仿/异戊醇提取纯培养菌株的基因组dna;
5)特异性引物扩增纯菌株的16srdna片段:采用特异性引物acido_31f:5‘-gatcctggctcagaatc-3’,acido_1492r:5‘-ggttaccttgttacgactt-3’,以步骤4中提取得到的纯培养菌株的基因组dna为模板进行pcr扩增。扩增后的产物经由2%的琼脂糖凝胶电泳检测,若成功得到阳性pcr产物且扩增片段大小为1462bp,即可证明筛选得到酸杆菌,若未获得阳性pcr产物则证明未筛选得到酸杆菌(原因可能为土壤中酸杆菌含量过低,致使筛选未成功)。
所述的改良后的液体基本培养基的配置方法为:kh2po41.5g,mgso47h2o0.2g,蔗糖30g,酵母提取物50mg(配置改良后的固体基本培养基时再添加琼脂粉20g,液体培养基无需添加琼脂粉),用蒸馏水定容至1000ml,采用1mol/l的hcl溶液调节ph至3.5-4。
改良后的固体基本培养基:kh2po41.5g,mgso47h2o0.2g,蔗糖30g,酵母提取物50mg,琼脂粉20g,用蒸馏水定容至1000ml,采用1mol/l的hcl溶液调节ph至3.5-4。
所述步骤4中所提取的纯培养菌株基因组dna:将步骤3中经分离纯化后的菌落接种改良后的液体基本培养基,以收集酸杆菌菌体。于25℃条件下,150rpm振荡过夜培养,8000rpm离心5min收集菌体,弃去上清液;加入tes缓冲液冲洗菌体,而后将菌体溶于20ml的tes缓冲液(tris-hcl50mm,edta5mm,nacl50mm,调节ph至8.0);加入50mg溶菌酶(终浓度2mg/ml),于37℃条件下孵育30min,孵育过程中每隔10min上下颠倒混匀一次;加入1.6ml的20%sds(终浓度2%),于60℃条件下水浴30min;加入3.6ml的5m次氯酸钠(naclo4)(终浓度1m)充分混匀;加入相同体积的的氯仿:异戊醇混合溶液(体积配比为24:1),上下颠倒混匀使液体呈现乳白色浑浊状,而后8000rpm(4℃)离心10min;取上清液置于灭菌后的烧杯中,加入等体积的预冷的异丙醇,将析出的dna丝状沉淀用灭菌的玻璃棒搅起而后溶于10mltes缓冲液中;加入50μl10mg/mlrnasea(终浓度50μg/ml)在37℃下消化30min以去除rna对后续实验的影响;加入100μl的10mg/ml蛋白酶k(proteinasek)(终浓度100μg/ml)在37℃下消化2h;加入等体积的氯仿:异戊醇混合溶液(体积配比为24:1),充分混匀,待其乳化后在4℃条件下,8000rpm离心10min;将上清液转移至一灭菌后的洁净烧杯中,加入等体积预冷的异丙醇和1/10体积的3mch3coona,用玻璃棒将析出的dna丝挑起;而后采用浓度为70%的乙醇溶液脱盐5min,空气中自然晾干以使酒精挥发干净;将缠有dna丝的玻璃棒放在3-5ml双蒸水(ddh2o)(含20μg/ml的rnasea)中,4℃孵育至dna全部溶解。dna于-20℃保存。
所述步骤5中pcr扩增所涉及的反应体系如下:1μldna模板,10×pcr反应缓冲液5μl,2.5mmol/ldntp混合溶液4μl,20pmol/l引物sph-429f1μl,20pmol/l引物sph-933r1μl,taq酶(5u/μl)0.25μl,加灭菌去离子水至总体积为50μl;pcr反应条件:95℃预变性4min,95℃变性40s,55℃退火40s,72℃延伸90s,35个循环,最后72℃延伸10min,pcr结束后的保存条件为4℃。
本发明检测原理:
酸杆菌门水平菌株16srdna中碱基序列号为31至1492的片段,该片段共含有1462个碱基
采用本发明设计的酸杆菌特异性引物进行pcr,能够特异性扩增该门水平下菌株16srdna的片段,因此获得pcr产物片段长度为1462的片段,即可以作为该菌株是否为酸杆菌的标准。
本发明的有益效果如下:
1.本发明结合了现代分子生物学技术和微生物学中的传统筛选培养基培养,提供特异性引物-pcr结合改良后的基本培养基平板特异性分离酸杆菌的方法,从环境样品中高效且特异性筛选并分离纯化酸杆菌,克服了以往获得酸杆菌所存在的困难。
2.采用本发明提供的特异性引物-pcr结合改良后的基本培养基平板特异性分离酸杆菌的方法,本实验所设计的引物对扩增酸杆菌16srdna具有强特异性,极大程度上降低了对其他微生物16srdna的错误扩增,相比于其他引物更为高效,并结合改良后的基本培养基,通过模拟酸杆菌的最适生活条件以高效筛选酸杆菌,同时抑制其他微生物类群的增殖以减少养分的争夺,最大程度的提高了酸杆菌的筛选效率。本发明可以直接从土壤样品中快速分离培养酸杆菌菌株资源,并在此基础上研究这些菌株的功能,有望获得携带特定功能基因(降解纤维素相关基因)的菌株。
3.实验室研究表明,采用本发明提供的特异性引物-pcr结合改良后的基本培养基平板的方法,能够土壤中直接筛选培养酸杆菌菌株,从而为进一步研究这些菌株的生态学功能和开发其潜在的其他价值提供了菌种资源。
附图说明
图1:土壤上清液稀释至原浓度10-4倍后的涂布平板结果;
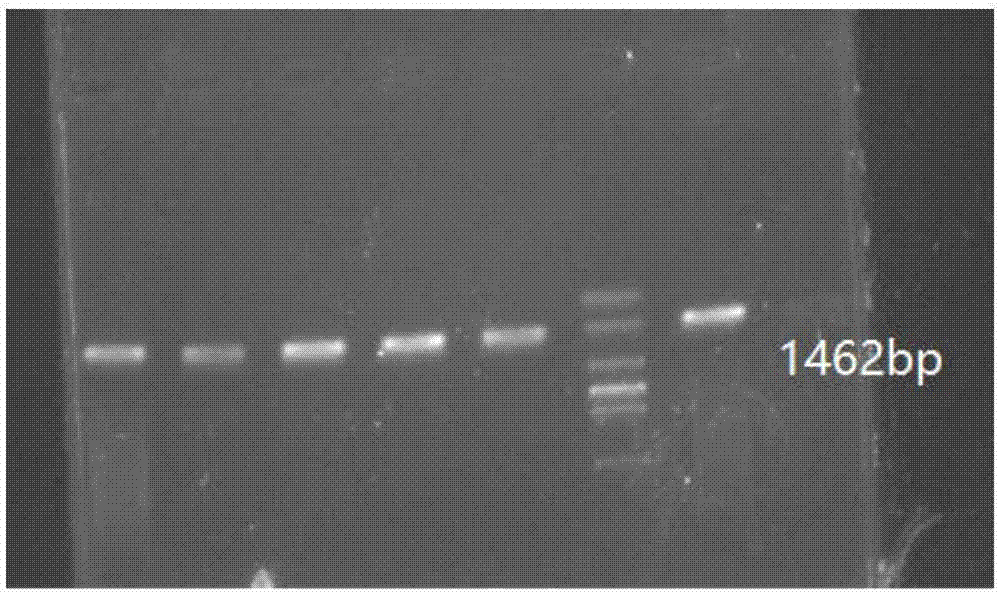
图2:采用本发明中的特异性引物对纯化菌株dna的pcr扩增结果;
图3:应用mega5对本方法筛选获得的酸杆菌菌株与genebank已确认的酸杆菌的16srdna构建的系统发育树。
具体实施方式
本发明筛选路线为:土壤样品采集→土壤微生物悬液制备→涂布改良的基本培养基平板→挑取白色单菌落连续划线分离纯化→提取纯菌株基因组dna→特异性引物pcr扩增16srdna片段→细菌通用引物扩增酸杆菌阳性菌株16srdna全长→测序验证。
详细实施步骤为:
1)土壤样品采集:土壤样品的采集深度为0-10cm,过2mm筛去除土壤中含有的石砾和细跟,4℃冰箱保存;
2)制备土壤微生物悬液:选取5g土壤样品置于已灭菌的50ml锥形瓶中并加入少量玻璃珠,而后加入45ml生理盐水,150rpm,振荡时间为2h。振荡后静置约30ming,用生理盐水对上清液进行10倍梯度稀释;
3)涂布改良后的基本培养基固体平板:取0.1ml步骤2)中稀释度为10-4的土壤悬液涂布至改良后的固体基本培养基平板上,30℃倒置培养3~6天;
4)连续划线分离纯化菌株:酸杆菌在改良后的固体基本培养基平板上多为白色单菌落(如附图1所示),因此挑取在改良后的固体基本培养基平板上生长的白色菌落,在改良后的固体基本培养基平板上反复划线分纯,直至分离出单菌落;
5)提取纯菌株基因组dna:将经分离纯化后的菌落接种改良后的液体基本培养基,首先收集酸杆菌菌体,于25℃条件下,150rpm振荡过夜培养,8000rpm离心5min收集菌体,弃去上清液;加入tes缓冲液冲洗菌体,而后将菌体溶于20ml的tes缓冲液;加入50mg溶菌酶(终浓度2mg/ml),于37℃条件下孵育30min,孵育过程中每隔10min上下颠倒混匀一次;加入1.6ml的20%sds(终浓度2%),于60℃条件下水浴30min;加入3.6ml的5m次氯酸钠(naclo4)(终浓度1m)充分混匀;加入相同体积的的氯仿:异戊醇混合溶液(体积配比为24:1),上下颠倒混匀使液体呈现乳白色浑浊状,而后8000rpm(4℃)离心10min;取上清液置于灭菌后的烧杯中,加入等体积的预冷的异丙醇,将析出的dna丝状沉淀用灭菌的玻璃棒搅起而后溶于10mltes缓冲液中;加入50μl10mg/mlrnasea(终浓度50μg/ml)在37℃下消化30min以去除rna对后续实验的影响;加入100μl的10mg/ml蛋白酶k(proteinasek)(终浓度100μg/ml)在37℃下消化2h;加入等体积的氯仿:异戊醇混合溶液(体积配比为24:1),充分混匀,待其乳化后在4℃条件下,8000rpm离心10min;将上清液转移至一灭菌后的洁净烧杯中,加入等体积预冷的异丙醇和1/10体积的3mch3coona,用玻璃棒将析出的dna丝挑起;而后采用浓度为70%的乙醇溶液脱盐5min,空气中自然晾干以使酒精挥发干净;将缠有dna丝的玻璃棒放在3-5ml双蒸水(ddh2o)(含20μg/ml的rnasea)中,4℃孵育至dna全部溶解。dna于-20℃保存。
6)酸杆菌16srdna特异性引物设计:通过ncbi公布确认的酸杆菌16srdna基因序列。采用mega5.0软件对选取的酸杆菌16srdna基因序列进行比对,然后通过plotcon软件进一步分析比对后的序列,确定序列的可变区和保守区,引物序列应位于保守区。对于初步选定的上下游引物质量评价均由软件primerpremier5完成。应用rdpⅱ数据库的sequencematch和ncbi数据库的blastsearch程序验证引物的特异性。最后选择一对最优的酸杆菌特异性引物acido_31f:5‘-gatcctggctcagaatc-3’,acido_1492r:5‘-ggttaccttgttacgactt-3’。
7)特异性引物扩增纯菌株16srdna片段:采用步骤6中设计的酸杆菌特异性引物acido_31f/acido_1492r,步骤5)中提取的基因组dna为模板pcr扩增步骤4)所分纯菌株的16srdna片段;50μl的pcr反应体系组成如下:2μldna模板,pcrmastermixture12.5μl,引物acdio_31f1μl(20pmol/l)引物acido_1492r1μl(20pmol/l),taq酶(5u/μl)0.25μl,甘油5μl,加灭菌去离子水至总体积为50μl;pcr反应条件:95℃预变性4min,95℃变性40s,55℃退火40s,72℃延伸90s,35个循环,最后72℃延伸10min,pcr结束后的保存条件为4℃。pcr反应的产物用1.5%琼脂糖凝胶电泳检测,扩增片段大小应为酸杆菌16srdna片段长度1462bp(如附图2所示)。
8)对可能的酸杆菌阳性菌株进行测序验证:选取步骤7中鉴定为酸杆菌的不同菌株,采用细菌通用引物515f/907r扩增16srdna全长序列,引物序列分别为,515f:5’-gtgccagcmgccgcgg-3’和907r:5’-ccgtcaattcmtttragttt-3’。所采用的pcr反应体系同步骤7);pcr扩增pcr反应体系组成如下:2μldna模板,pcrmastermixture12.5μl(美国,promega公司),引物acdio_31f1μl(20pmol/l)引物acido_1492r1μl(20pmol/l),taq酶(5u/μl)0.25μl,甘油5μl,加灭菌去离子水至总体积为50μl;pcr反应条件:95℃预变性4min,95℃变性40s,55℃退火40s,72℃延伸90s,35个循环,最后72℃延伸10min,pcr结束后的保存条件为4℃。,pcr结束后的保存条件为4℃。pcr产物经凝胶回收纯化后,委托商业测序公司测序。将所得序列在genbank数据库进行blast比对分析,获得相似序列。应用clustalx软件将测序序列与相似序列比对,然后用mega5.0软件构建系统发育树。若与已被鉴定的酸杆菌相似性达到97%以上,则可认为其为酸杆菌。
实施例1
采集我国江西省武夷山森林土壤,过2mm筛,4℃冰箱保存。称取5g土样置于已灭菌的装有玻璃珠的锥形瓶中,加入45ml生理盐水,于150rpm振荡2h。而后将悬浮液静置,取上清液,用生理盐水对土壤悬液进行10倍体积梯度稀释。分别取0.1ml稀释度为10-3、10-4、10-5、10-6的土壤悬液涂布至添加改良后的固体基本培养基平板上,30℃倒置培养3~6天。挑取改良后的固体基本培养基平板上生长的白色菌落(图1),在改良后的固体基本培养基平板上反复划线分纯,直至分离出单菌落。
所述的改良后的液体基本培养基的配置方法为:kh2po41.5g,mgso47h2o0.2g,蔗糖30g,酵母提取物50mg,琼脂粉20g(配置改良后的固体基本培养基时添加,液体培养基无需添加琼脂粉),用蒸馏水定容至1000ml,采用1mol/l的hcl溶液调节ph至3.5-4。
将上述分纯的9个白色单菌落,接种于3ml改良后的液体基本培养基中,25℃摇床过夜培养,采用sds、蛋白酶k和氯仿/异戊醇方法提取基因组dna,并采用dna凝胶回收试剂盒2.0进行纯化(宝生物工程(大连)有限公司,中国大连))。采用酸杆菌特异性引物acido_31f/1492r通过pcr扩增16srdna片段,50μl的pcr反应体系组成如下:1;50μl的pcr反应体系组成如下:1μldna模板,10×pcr反应缓冲液5μl,2.5mmoll-1dntp混合溶液4μl,20pmoll-1引物sph-429f1μl,20pmoll-1引物sph-933r1μl,taq酶(5u/μl)0.25μl,加灭菌去离子水至总体积为50μl。pcr反应条件:94℃预变性5min,94℃变性1min,55℃退火45s,72℃延伸90s,35个循环,最后72℃延伸8min,pcr结束后的保存条件为4℃。pcr反应的产物用2%琼脂糖凝胶电泳检测(见图2),pcr产物片段大小为1462bp,与酸杆菌16srdna片段相同,即样品中存在酸杆菌,符合要求的为7个菌株。
对上述7个菌株扩增16srdna序列的blastn分析表明,均归属为酸杆菌,具体为,菌株strain1与unculturedacidobacteriabacteriumcloned-16s-96gu552204.1(gu552204.1)相似性最高(98%);菌株strain2与unculturedacidobacteriabacteriumclonerc2-3(ef626824.1)的同源性最高(99%);菌株strain3与unculturedacidobacteriabacteriumclone91(eu097143.1)亲缘关系最近(100%);菌株strain5与unculturedacidobacteriabacteriumclonef155cmcontigb1(jn003098.1)同源性最高(99%);菌株6与unculturedacidobacteriabacteriumcloned-16s-17(eu603382.1)同源性最高(99%),菌株7与unculturedacidobacteriabacteriumcloned-16s-84(gu552194.1)同源性最高(98%)。
采用mega5.0软件对上述酸杆菌的标准菌株序列进行比对,并构建系统发育树(图3)。
对比例
为对比验证本发明中的改良后的基本培养基,采用普通基础培养基为对照组进行实验,所用样品与实施例1中相同,相关操作如土壤采集、土壤悬液的制备与稀释均与实施例1一致,而后分别取0.1ml稀释度为10-3、10-4、10-5、10-6的土壤悬液涂布至普通固体基本培养基平板上,30℃倒置培养3~6天后,普通固体基本培养基上生长有多种表型不一致的菌落,不存在筛选效果,对比证明本方法中的改良后的基本培养基可有效筛选出酸杆菌菌株。
应用例
采集我国江西武夷山地区森林土壤(0-10cm)作为供试土壤。土壤悬液涂布改良后的固体基本培养基平板。从平板上共随机挑取白色单菌落,经反复划线分纯。提取这9个单菌落的基因组dna,采用用酸杆菌-特异性引物进行pcr扩增。在9个单菌落中,有7个白色单菌落产生片段大小合适的pcr产物。将上述7个pcr扩增片段进行测序。采用细菌通用引物515f/907r扩增这些菌株的16srdna全长序列,pcr产物纯化后进行测序,且blast比对也表明筛选获得的微生物菌株均为酸杆菌。
sequencelisting
<110>中国科学院沈阳应用生态研究所
<120>一种从土壤中特异性分离酸杆菌的方法
<130>
<160>4
<170>patentinversion3.5
<210>1
<211>17
<212>dna
<213>人工序列
<220>
<221>gene
<222>(1)..(17)
<400>1
gatcctggctcagaatc17
<210>2
<211>19
<212>dna
<213>人工序列
<220>
<221>gene
<222>(1)..(19)
<400>2
ggttaccttgttacgactt19
<210>3
<211>16
<212>dna
<213>人工序列
<220>
<221>gene
<222>(1)..(16)
<400>3
gtgccagcmgccgcgg16
<210>4
<211>20
<212>dna
<213>人工序列
<220>
<221>gene
<222>(1)..(20)
<400>4
ccgtcaattcmtttragttt20
- 还没有人留言评论。精彩留言会获得点赞!