一种吲哚并咔唑共价有机框架材料及其合成方法与流程
本发明涉及有机合成及功能材料领域,具体涉及一种吲哚并咔唑共价有机框架材料及其合成方法。
背景技术:
吲哚并咔唑具有较大的平面共轭结构,有利于电子转移,而且它也具有很强的结构可调性。以吲哚[3,2-b]咔唑为例,我们可以将其很多位置上的氢用不同的官能团取代,得到具有不同光电性能和机械性能的吲哚并咔唑衍生物,例如将5,11-位置上的氢用脂肪链取代,能够增加吲哚并咔唑的溶解度;在2,8-和3,9-位上的取代能够提高其分子堆积,而且在这些可取代的位置引入特定的基团能够有利于电荷载流子迁移和载流子的注入,使得其合成的寡聚物和高分子都具有独特的电学、光学、磁学等方面的性质。因其结构的独特性,吲哚并咔唑被广泛应用于药物分子、有机发光二极管、有机场效应晶体管、太阳能电池、传感器等方面。
共价有机框架材料是一种新兴的类分子筛材料,在近年来引起了广泛的关注,目前在气体吸附/分离/储存,药物缓释,光电(器件),催化等领域已有初步的应用。共价有机框架材料具有大π共轭体系和晶型的结构,使得它在光电和传感领域具有很好的应用前景。
技术实现要素:
基于上述现有技术的现状,本发明的目的是提供一种吲哚并咔唑共价有机框架材料及其合成方法。
具体地,本发明提供一种吲哚并咔唑共价有机框架材料的合成方法,其特征在于,将均苯三甲醛与吲哚并咔唑前体在有机溶剂中混合均匀后,在醋酸的催化下反应得到吲哚并咔唑共价有机框架材料;
所述吲哚并咔唑前体为下述式1、式2或式3所示化合物;
式中,n-bu表示正丁基。
其中,所述有机溶剂为均三甲苯、1,4-二氧六环、或者1,4-二氧六环与均三甲苯的混合液、乙醇与均三甲苯的混合液、四氢呋喃与均三甲苯的混合液。
此外,优选所述有机溶剂为均三甲苯、1,4-二氧六环、或者体积比为1:4-4:1的1,4-二氧六环与均三甲苯的混合液、体积比为1:4-4:1的乙醇与均三甲苯的混合液、体积比为1:4-4:1的四氢呋喃与均三甲苯的混合液。
此外,均苯三甲醛与吲哚并咔唑前体的总浓度为1-100g/l,其中均三苯甲醛与吲哚并咔唑前体的摩尔比为4:6。
此外,醋酸的用量为均苯三甲醛摩尔用量的0.8-40倍。
此外,反应在80-150℃下进行。
此外,上述的反应在密封的反应器中发生,反应时间为3天。离心后干燥得到吲哚并咔唑共价有机框架材料。
本发明还提供一种如上所述的合成方法制得的吲哚并咔唑共价有机框架材料。
通过材料在x射线衍射谱图中的几个明显的衍射峰可知,材料的结构是长程有序的,而材料的孔径分布曲线说明其具有规则的孔道结构,再结合拓扑学知识和计算机模拟可知:通过本发明所述的合成方法制得的手性共价有机框架材料具有长程有序的二维六方结构和规则的孔道。
附图说明
图1是实施例1中产物及原料的x射线衍射谱图。
图2是实施例2中产物及原料的x射线衍射谱图。
图3是实施例3中产物及原料的x射线衍射谱图。
图4是实施例1-3中产物的固体核磁谱图。
图5是实施例1中产物及原料的红外谱图。
图6是实施例2中产物及原料的红外谱图。
图7是实施例3中产物及原料的红外谱图。
图8实施例1-3中产物的氮气吸脱附曲线。
图9是实施例1-3中产物的孔径分布曲线。
图10是实施例1-3中产物的热重曲线,其中曲线a)对应于iczcof-lzu171、曲线b)对应于iczcof-lzu172、曲线c)对应于iczcof-lzu170。
具体实施方式
以下结合实施例对本发明进一步说明,应当理解,此处所描述的优选实施例仅用于说明和解释本发明,并不用于限定本发明。
(一)吲哚并咔唑前体的合成方法
(1)吲哚并咔唑前体1的合成方法如下:
化合物5的合成
将乙酸钠(33.9g,0.414mmol)的水溶液(100ml)加入3-溴苯肼盐酸盐的乙醇溶液(200ml)中,室温搅拌15min,将1,4-环己二酮的乙醇溶液(50ml)加入体系之后再加入乙酸(50ml)。将体系在50℃下反应1h,再降至0℃反应1h,抽滤,大量水洗,晾干,得到白色固体4。在10℃下将得到化合物4分批加入硫酸/乙酸(15ml/75ml)的混合酸中,并保持在10℃下搅拌10min,再升至室温搅拌10min,然后升至65℃反应后搅拌15min,最后降至室温并搅拌过夜。反应结束后抽滤,滤饼加入到甲醇(200ml)中,回流30min,抽滤,滤饼用dmf重结晶三次,最后得到黄绿色固体5(1.7g).1hnmr(400mhz,dmso-d6)δ,ppm:11.3(s,2h),8.5(d,2h),8.2(s,2h),7.5(dd,2h),7.4(d,2h).
化合物6的合成
将化合物5(1.68g,4mmol)、n-bubr(2.19g,16mmol)、苄基三乙基氯化铵(180mg,0.8mmol)加入100ml两颈烧瓶中,加入dmso(40ml)后,再将50%naoh水溶液(8ml)加入,室温搅拌1h,再在50℃下搅拌4h。反应结束后,倒入400ml甲醇溶液中,抽滤,分别用水,dmf,甲醇,丙酮各洗3次,真空干燥后得到6.1hnmr(400mhz,cdcl3)δ,ppm:8.3(d,2h),8.0(s,2h),7.6(dd,2h),7.3(d,2h),4.4(t,4h),2.0(m,4h),1.5(m,4h),1.0(t,6h).
吲哚并咔唑前体1的合成
将binap(380mg,0.6mmol)、pd2(dba)3(280mg,0.3mmol)加入200ml单口烧瓶,氩气置换气三次,再将已冷冻脱换气三次的甲苯(100ml)加入,110℃下回流30min。冷却至室温后,加入6(1.58g,3mmol)、ph2c=nh(2ml,12mmol)、t-buona(1.15g,12mmol),再次冷冻脱气三次,然后110℃回流过夜。反应结束后,直接上样,柱层析分离,收集橙黄色液体,减压浓缩,将所得固体加入100ml单口烧瓶,氩气置换气三次,氩气保护下加入干燥的thf(40ml),再在冰浴条件下滴加2m盐酸(20ml),室温搅拌3-5h。tlc检测反应结束后加入3mnaoh(20ml),室温搅拌30min,再用dcm萃取,收集有机相并用无水na2so4干燥,浓缩有机相,柱层析分离,得吲哚并咔唑前体1.1hnmr(400mhz,cdcl3)δ,ppm:7.9(d,2h),7.8(s,2h),6.7(d,2h),6.6(dd,2h),4.3(t,4h),2.0(m,4h),1.5(m,4h),1.0(t,6h).13cnmr(400mhz,cdcl3)δ,ppm:145.0,143.1,136.0,121.8,120.7,115.8,107.4,97.4,94.2,43.0,30.9,20.7,14.0.hrms:m/zcalcdforc26h30n4[m+h]+,399.2543;found,399.2533.
(2)吲哚并咔唑前体2的合成方法如下:
化合物7的合成
将nah(288mg,12mmol)溶于10mldmso中,再将溶于dmso(10ml)的化合物5(818mg,2mmol)缓慢加入,室温下搅拌0.5h。再将溶于dmso(10ml)的二甲氨基乙烷盐酸盐(634mg,4.4mmol)缓慢滴入,氩气保护下室温条件下搅拌4h。反应结束后,将体系倒入冰水(200ml)中搅拌1h,抽滤,用水和乙酸乙酯洗涤,烘干,得黄色固体7.1hnmr(400mhz,cdcl3)δ,ppm:8.0(d,2h),7.9(s,2h),7.6(d,2h),7.4(dd,2h),4.5(t,4h),2.8(t,4h),2.4(s,12h).
吲哚并咔唑前体2的合成
将binap(380mg,0.6mmol)、pd2(dba)3(280mg,0.3mmol)加入200ml单口烧瓶,氩气置换气三次,再将已冷冻脱换气三次的甲苯(100ml)加入,110℃下回流30min。冷却至室温后,加入7(1.67g,3mmol)、ph2c=nh(2ml,12mmol)、t-buona(1.15g,12mmol),再冷冻脱气三次,然后110℃回流过夜。反应结束后,直接上样,柱层析分离,收集橙黄色液体,减压浓缩,将所得固体加入100ml单口烧瓶,氩气置换气三次,氩气保护下加入干燥的thf(40ml),再在冰浴条件下滴加2m盐酸(20ml),室温搅拌3-5h。tlc检测反应结束后加入3mnaoh(20ml),室温搅拌30min,再用dcm萃取,收集有机相并用无水na2so4干燥,浓缩有机相,柱层析分离,得吲哚并咔唑前体2.1hnmr(400mhz,cdcl3)δ,ppm:7.9(d,2h),7.8(s,2h),6.7(d,2h),6.6(dd,2h),4.4(t,4h),2.8(t,4h),2.4(s,12h).13cnmr(400mhz,cdcl3)δ,ppm:148.0,143.3,135.7,121.6,120.9,113.7,107.6,97.4,93.0,57.2,46.0,41.3.hrms:m/zcalcdforc26h32n6[m+h]+,429.2761;found,429.2755.
(3)吲哚并咔唑前体3的合成方法如下:
化合物9的合成
将6-溴吲哚(4.95g,25.0mmol)和苯甲醛(2.53ml,25.0mmol)加入250ml三口烧瓶中,氩气置换气三次,氩气保护下加入ch3cn(140ml),再在冰浴条件下滴加45%hi(0.95ml,5.0mmol),最后在80℃下回流14h。tlc检测反应完全,将体系冷却至室温,抽滤并用冷的ch3cn洗涤,烘干,得8和9的混合物4.1g。将所得到的混合物和i2(2.03g,8.0mmol)加入装有磁子的250ml单口烧瓶中,氩气置换气三次,氩气保护下加入ch3cn(130ml),再在80℃下回流14h。tlc检测反应完全,将体系减压浓缩至大约30ml,抽滤并用冷的ch3cn洗涤,烘干,得3.36g黄色固体9,直接用于下步合成。
化合物10的合成
将化合物9(3.36g,5.9mmol)加入装有磁子的100ml两颈烧瓶中,氩气置换气三次,氩气保护的条件下加入干燥的dmf(30ml),冰浴条件下分批加入nah(0.57g,23.7mmol),再升至室温,搅拌1h。再在冰浴条件下滴加n-bubr(2.6ml,23.7mmol),室温搅拌14h。tlc检测反应结束,将体系倒入50%乙醇水溶液(80ml),搅拌1h后抽滤,乙醇洗涤,再烘干,得化合物10,直接用于下步合成。
吲哚并咔唑前体3的合成
将binap(75mg,0.12mmol)、pd2(dba)3(55mg,0.06mmol)加入100ml两颈烧瓶,氩气置换气三次,再将已冷冻脱换气3次的甲苯(20ml)加入,110℃下回流30min。冷却至室温后,加入10(407mg,0.6mmol)、ph2c=nh(0.4ml,2.4mmol)、t-buona(231mg,2.4mmol),再次冷冻脱气三次,然后110℃回流过夜。反应结束后,直接上样,收集橙黄色液体,减压浓缩,将所得固体加入100ml单口烧瓶,氩气置换气三次,氩气保护下加入干燥的thf(20ml),再在冰浴条件下滴加浓盐酸(10ml),室温搅拌。tlc检测反应结束后滴加氨水至ph=10左右,旋干体系中的thf,再用dcm萃取,收集有机相并用无水na2so4干燥,浓缩有机相,柱层析分离,得吲哚并咔唑前体3.1hnmr(400mhz,cdcl3)δ,ppm:7.7(m,4h),7.6(m,6h),6.5(d,2h),6.2(m,4h),3.7(t,4h),1.5(m,4h),1.0(m,4h),0.9(t,6h).13cnmr(400mhz,cdcl3)δ,ppm:144.6,143.9,139.3,132.2,130.7,128.8,127.8,122.9,121.7,116.4,115.9,107.5,93.9,44.1,30.6,20.0,13.8.hrms:m/zcalcdforc38h38n4[m+h]+,551.3169;found,551.3161.
(二)吲哚并咔唑共价有机框架材料的合成:
实施例1
取一10ml玻璃封管,加入6.5mg均三苯甲醛和23.9mg吲哚并咔唑前体1,再加入二氧六环(1.0ml)并摇匀,最后缓慢滴加0.2ml3m的醋酸水溶液。再用液氮将封管冷冻,在真空吸附线下将玻璃封管内压力抽至0mbar,然后用火焰枪将瓶颈封住。待体系升至室温后,放入100℃的烘箱中反应3天。反应结束后冷却至室温,小心将瓶颈敲碎,将生成的固体转移到离心管中,依次用thf和丙酮各洗三次,再用无水乙醚洗一次,自然干燥,得到黄色固体粉末iczcof-lzu171。
实施例2
取一10ml玻璃封管,加入6.5mg均三苯甲醛和25.7mg吲哚并咔唑前体2,再加入二氧六环(1.0ml)并摇匀,最后缓慢滴加0.2ml3m的醋酸水溶液。再用液氮将封管冷冻,在真空吸附线下将玻璃封管内压力抽至0mbar,然后用火焰枪将瓶颈封住。待体系升至室温后,放入100℃的烘箱中反应3天。反应结束后冷却至室温,小心将瓶颈敲碎,将生成的固体转移到离心管中,依次用thf和丙酮各洗三次,再用无水乙醚洗一次,自然干燥,得到黄色固体粉末iczcof-lzu172。
实施例3
取一10ml玻璃封管,加入4.9mg均三苯甲醛和24.8mg吲哚并咔唑前体3,再加入均三甲苯(1.0ml)并摇匀,最后缓慢滴加0.2ml3m的醋酸水溶液。再用液氮将封管冷冻,在真空吸附线下将玻璃封管内压力抽至0mbar,然后用火焰枪将瓶颈封住。待体系升至室温后,放入100℃的烘箱中反应3天。反应结束后冷却至室温,小心将瓶颈敲碎,将生成的固体转移到离心管中,依次用thf和丙酮各洗三次,再用无水乙醚洗一次,自然干燥,得到棕黄色固体粉末iczcof-lzu170。
图1是实施例1中产物及原料的x射线衍射谱图。
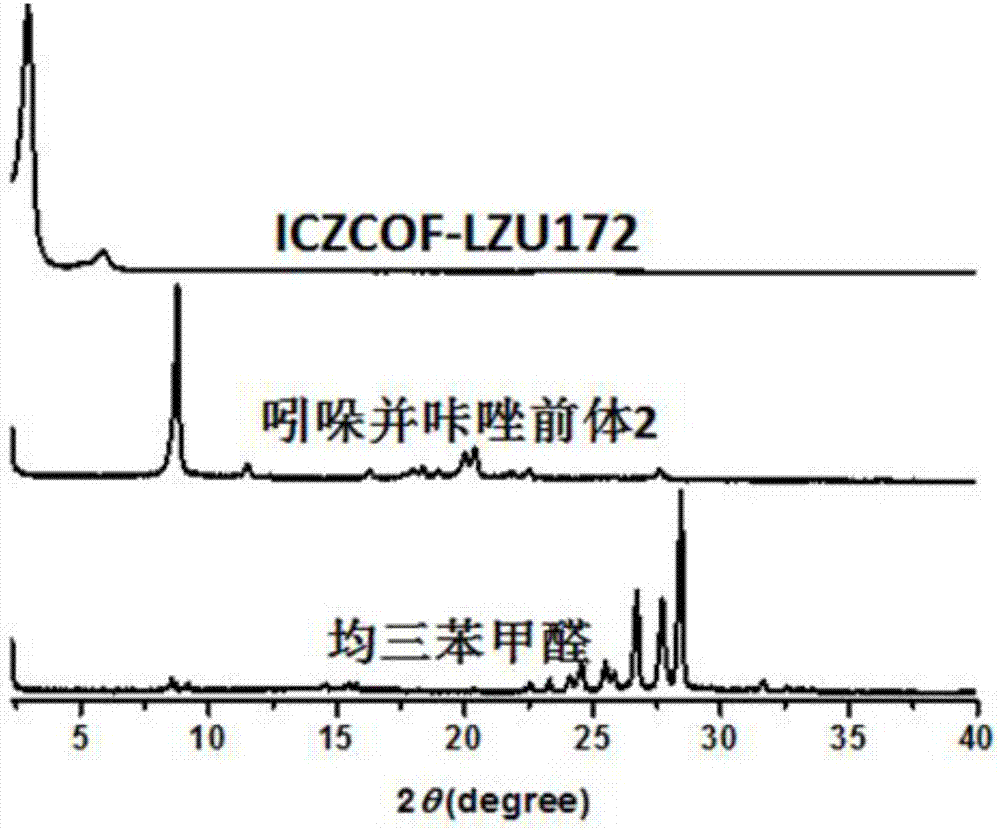
图2是实施例2中产物及原料的x射线衍射谱图。
图3是实施例3中产物及原料的x射线衍射谱图。
图4是实施例1-3中产物的固体核磁谱图。
图5是实施例1中产物及原料的红外谱图。
图6是实施例2中产物及原料的红外谱图。
图7是实施例3中产物及原料的红外谱图。
图8实施例1-3中产物的氮气吸脱附曲线。
图9是实施例1-3中产物的孔径分布曲线。
图10是实施例1-3中产物的热重曲线,其中曲线a)对应于iczcof-lzu171、曲线b)对应于iczcof-lzu172、曲线c)对应于iczcof-lzu170。
通过图1-3中iczcof-lzu171、iczcof-lzu172、iczcof-lzu170与各自原料的粉末x射线衍射图谱的对比,可以确定本发明成功的合成了三例新的晶型材料。
图8及图9为各个产物的氮气吸脱附曲线以及孔径分布曲线,该数据表明该材料具有较大的比表面积和规则的孔道结构。
图10为材料的热重曲线,其中曲线a)对应于iczcof-lzu171,曲线b)对应于iczcof-lzu172,曲线c)对应于iczcof-lzu170。
最后应说明的是:以上所述仅为本发明的优选实施例而已,并不用于限制本发明,尽管参照前述实施例对本发明进行了详细的说明,对于本领域的技术人员来说,其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!