定量检测重组慢病毒的滴度的方法与流程
本发明涉及生物检测领域,特别是涉及一种定量检测重组慢病毒的滴度的方法。
背景技术:
重组慢病毒(lentivirus)载体是以hiv-1(人类免疫缺陷i型病毒)为基础发展起来的基因治疗载体,对分裂细胞和非分裂细胞均具有感染能力,是目前应用最广泛的基因运载工具之一,广泛应用于转基因动物的制备和基因治疗研究中。
重组慢病毒制备的过程中,通常用病毒的滴度来衡量重组慢病毒的制备是否成功以及重组慢病毒的质量,因此滴度测定是一个重要的步骤。现有技术中,测定重组慢病毒滴度的主要方法有流式细胞术(facs)、酶联免疫(elisa)法、金标试纸法和定量pcr法。
其中,facs法是最为准确的测定重组慢病毒滴度的方法。该法先用病毒液感染靶细胞(通常为293t细胞),之后通过流式细胞术测定其中被成功感染的细胞的比例。该方法虽然可准确测定重组慢病毒的实际感染能力,所测滴度数据能准确反映病毒的实际感染能力,但该方法需要重组慢病毒载体表达荧光标记基因(如绿色荧光蛋白gfp),对缺乏此类标记基因的重组慢病毒则无能为力。
elisa法和金标试纸法是定量检测的都是重组慢病毒颗粒中p24蛋白的表达水平,从而推算病毒滴度。在实际情况中,由于病毒液里总是含有一定的游离p24蛋白,因此通过elisa法测定的重组慢病毒滴度通常都会偏高;金标试纸法虽然操作简便,所需时间短,但它只能起到定性的目的,无法对慢病毒滴度进行准确定量测定。
传统的定量pcr法可测定病毒感染的靶细胞中平均每个细胞基因组中病毒基因组的拷贝数,可在一定程度上反应重组慢病毒的滴度。但是该方法虽然在无标记基因重组慢病毒的滴度测定中可行,但获得慢病毒基因组的拷贝数后其仍需依赖有荧光标记物的重组慢病毒作为对照才能准确计算待测样品中重组慢病毒的滴度,操作相对繁琐和不便。
综上,传统的检测方法均无法实现在不借助荧光标记物的条件下准确测定重组慢病毒的滴度。
技术实现要素:
基于此,有必要提供一种能够在不借助荧光标记物的条件下准确测定重组慢病毒的滴度的定量检测重组慢病毒的滴度的方法。
一种定量检测重组慢病毒的滴度的方法,包括以下步骤:
用待测样品感染靶细胞,并提取感染后所述靶细胞中的基因组dna,其中所述待测样品中含有重组慢病毒;
测定所述基因组dna中wpre元件的拷贝数以及htert基因的拷贝数;
根据所述wpre元件的拷贝数和所述htert基因的拷贝数计算平均每个靶细胞中重组慢病毒的颗粒数,其中,平均每个靶细胞中重组慢病毒的颗粒数=(所述wpre元件的拷贝数/所述htert基因的拷贝数)×2;及
根据所述靶细胞的总数和所述平均每个靶细胞中重组慢病毒的颗粒数计算单位体积的所述待测样品中含重组慢病毒的颗粒数,得到所述待测样品中重组慢病毒的滴度。
在一个实施方式中,所述测定所述基因组dna中wpre元件的拷贝数以及htert基因的拷贝数的步骤包括:
将所述基因组dna与wpre元件上游引物以及wpre元件下游引物混合进行实时荧光定量pcr扩增反应,获取扩增所述wpre元件的ct值;
将所述基因组dna与htert基因上游引物以及htert基因下游引物混合进行实时荧光定量pcr扩增反应,获取扩增所述htert基因的ct值;
将扩增所述wpre元件的ct值带入由wpre元件标准品建立的标准曲线中,获得所述基因组dna中所述wpre元件的含量;
将扩增所述htert基因的ct值带入由htert基因标准品建立的标准曲线中,获得所述基因组dna中所述htert基因的含量;及
根据所述wpre元件的含量计算所述基因组dna中所述wpre元件的拷贝数,以及根据所述htert基因的含量计算所述基因组dna中所述htert基因的拷贝数。
在一个实施方式中,所述wpre元件上游引物的碱基序列seqidno.1所示,所述wpre元件下游引物的碱基序列seqidno.2所示。
在一个实施方式中,所述htert基因上游引物的碱基序列seqidno.3所示,所述htert基因下游引物的碱基序列seqidno.4所示。
在一个实施方式中,所述由wpre元件标准品建立的标准曲线通过如下操作得到:
将wpre元件标准品进行梯度稀释,获得梯度浓度的所述wpre元件标准品;
将每一个浓度的所述wpre元件标准品分别与所述wpre元件上游引物以及所述wpre元件下游引物混合进行实时荧光定量pcr扩增反应,获取扩增每一个浓度的所述wpre元件标准品的ct值;及
根据所述wpre元件标准品的ct值与浓度的对应关系建立所述wpre元件标准品的标准曲线。
在一个实施方式中,所述由htert基因标准品建立的标准曲线通过如下操作得到:
将htert基因标准品进行梯度稀释,获得梯度浓度的所述htert基因标准品;
将每一个浓度的所述htert基因标准品分别与所述htert基因上游引物以及所述htert基因下游引物混合进行实时荧光定量pcr扩增反应,获取扩增每一个浓度的所述htert基因标准品的ct值;及
根据所述htert基因标准品的ct值与浓度的对应关系建立所述htert基因标准品的标准曲线。
在一个实施方式中,所述wpre元件标准品通过如下操作得到:将wpre元件模板与wpre元件模板的上游引物以及wpre元件模板的下游引物混合进行pcr扩增反应,收集所述pcr扩增反应的产物,得到所述wpre元件标准品,其中,所述wpre元件模板的上游引物的碱基序列seqidno.5所示,所述wpre元件模板的下游引物的碱基序列seqidno.6所示;及/或,
所述htert基因标准品通过如下操作得到:将htert基因模板与htert基因模板的上游引物以及htert基因模板的下游引物混合进行pcr扩增反应,收集所述pcr扩增反应的产物,得到所述htert基因标准品,其中,所述htert基因模板的上游引物的碱基序列seqidno.7所示,所述htert基因模板的下游引物的碱基序列seqidno.8所示。
在一个实施方式中,所述根据所述wpre元件的含量计算所述基因组dna中所述wpre元件的拷贝数,以及根据所述htert基因的含量计算所述基因组dna中所述htert基因的拷贝数的步骤中:
所述wpre元件的拷贝数=(na×所述wpre元件的含量)/所述wpre元件的标准分子量;
所述htert基因的拷贝数=(na×所述htert基因的含量)/所述htert基因的标准分子量;
其中,na表示阿伏伽德罗常数,所述wpre元件的标准分子量=一对碱基的平均分子量×所述wpre元件标准品的碱基对数,所述htert基因的标准分子量=一对碱基的平均分子量×所述htert基因标准品的碱基对数。
在一个实施方式中,所述靶细胞为293t细胞。
在一个实施方式中,所述根据所述靶细胞的总数和所述平均每个靶细胞中重组慢病毒的颗粒数计算单位体积的所述待测样品中含重组慢病毒的颗粒数的步骤中:
单位体积的所述待测样品中含重组慢病毒的颗粒数=(所述靶细胞的总数×所述平均每个靶细胞中重组慢病毒的颗粒数)/所述待测样品的体积。
上述定量检测重组慢病毒的滴度的方法,先用含重组慢病毒的待测样品感染靶细胞,并提取感染后靶细胞中的基因组dna,然后测定基因组dna中wpre元件的拷贝数以及htert基因的拷贝数。根据wpre元件的拷贝数和htert基因的拷贝数计算平均每个靶细胞中重组慢病毒的颗粒数。之后根据靶细胞的总数和平均每个靶细胞中重组慢病毒的颗粒数计算单位体积的待测样品中含重组慢病毒的颗粒数,即得到待测样品中重组慢病毒的滴度。其中,wpre元件是重组慢病毒中常见的调控元件,且在正常情况下不存在于动物细胞体内,因此可以作为重组慢病毒插入靶细胞基因组后的一个定量检测标志物。而htert基因(人端粒酶催化亚基基因)是细胞中一个稳定的单拷贝基因,htert基因的表达水平可作为内参。重组慢病毒感染靶细胞后,将包含wpre元件的整个外源基因表达框导入到靶细胞的基因组中,用wpre元件的拷贝数除以htert基因的拷贝数再乘以2(乘以2是因为靶细胞是二倍体,存在两个等位基因)即可获得平均每个靶细胞中重组慢病毒的颗粒数,即单个基因组中导入重组慢病毒基因的拷贝水平。然后根据靶细胞的总数和平均每个靶细胞中重组慢病毒的颗粒数即可计算单位体积内待测样品中含重组慢病毒的颗粒数,得到待测样品中重组慢病毒的滴度。发明人在标记基因的选择、测算方法等方面进行了大量的探索研究,预料不到地发现,通过测定感染重组慢病毒后的靶细胞的基因组dna中wpre元件的拷贝数以及htert基因的拷贝数即可准确测算重组慢病毒的滴度。上述定量检测重组慢病毒的滴度的方法操作简便,能够在不借助荧光标记物的条件下准确定量测定重组慢病毒的滴度。
附图说明
图1为一实施方式的定量检测重组慢病毒的滴度的方法的流程图;
图2为图1中测定基因组dna中wpre元件的拷贝数以及htert基因的拷贝数的方法的流程图;
图3为实施例1中wpre元件pcr产物和htert基因pcr产物的琼脂糖凝胶电泳比对图;
图4为实施例2中wpre元件标准品的扩增曲线图;
图5为实施例2中wpre元件标准品的熔解曲线图;
图6为实施例2中以log10为底wpre元件标准品的含量的对数为横坐标,以wpre元件标准品的ct值为纵坐标绘制的标准曲线图;
图7为实施例2中htert基因标准品的扩增曲线图;
图8为实施例2中htert基因标准品的熔解曲线图;
图9为实施例2中以log10为底htert基因标准品的含量的对数为横坐标,以htert基因标准品的ct值为纵坐标绘制的标准曲线图。
具体实施方式
为使本发明的上述目的、特征和优点能够更加明显易懂,下面结合具体实施例及附图对本发明的具体实施方式做详细的说明。在下面的描述中阐述了很多具体细节以便于充分理解本发明。但是本发明能够以很多不同于在此描述的其它方式来实施,本领域技术人员可以在不违背本发明内涵的情况下做类似改进,因此本发明不受下面公开的具体实施的限制。
请参阅图1,一实施方式的定量检测重组慢病毒的滴度的方法包括以下步骤s110~s140。
s110、用待测样品感染靶细胞,并提取感染后靶细胞中的基因组dna,其中待测样品中含有重组慢病毒。
具体地,待测样品为含有重组慢病毒的病毒悬液。
重组慢病毒中含有wpre元件(调控元件),若重组慢病毒成功感染靶细胞后,可将包含wpre元件的整个外源基因表达框导入到靶细胞的基因组中。
在一个实施方式中,靶细胞为293t细胞。用重组慢病毒感染293t细胞时,感染效率高,因此293t细胞非常适合作为重组慢病毒感染的靶细胞。
在一个实施方式中,用待测样品感染靶细胞,并提取感染后靶细胞中的基因组dna的步骤包括:复苏培养靶细胞1天~2天,向培养基中加入待测样品感染靶细胞,培养靶细胞1天~2天后更换新鲜培养基,继续培养2天~5天,收集感染后培养的靶细胞,并用试剂盒提取靶细胞中的基因组dna。
s120、测定s110中得到的基因组dna中wpre元件的拷贝数以及htert基因的拷贝数。
wpre元件是重组慢病毒中常见的调控元件,且在正常情况下不存在于动物细胞体内,因此可以作为重组慢病毒插入靶细胞基因组后的一个定量检测标志物。而htert基因是靶细胞中一个稳定的单拷贝基因,htert基因的表达水平可作为内参。
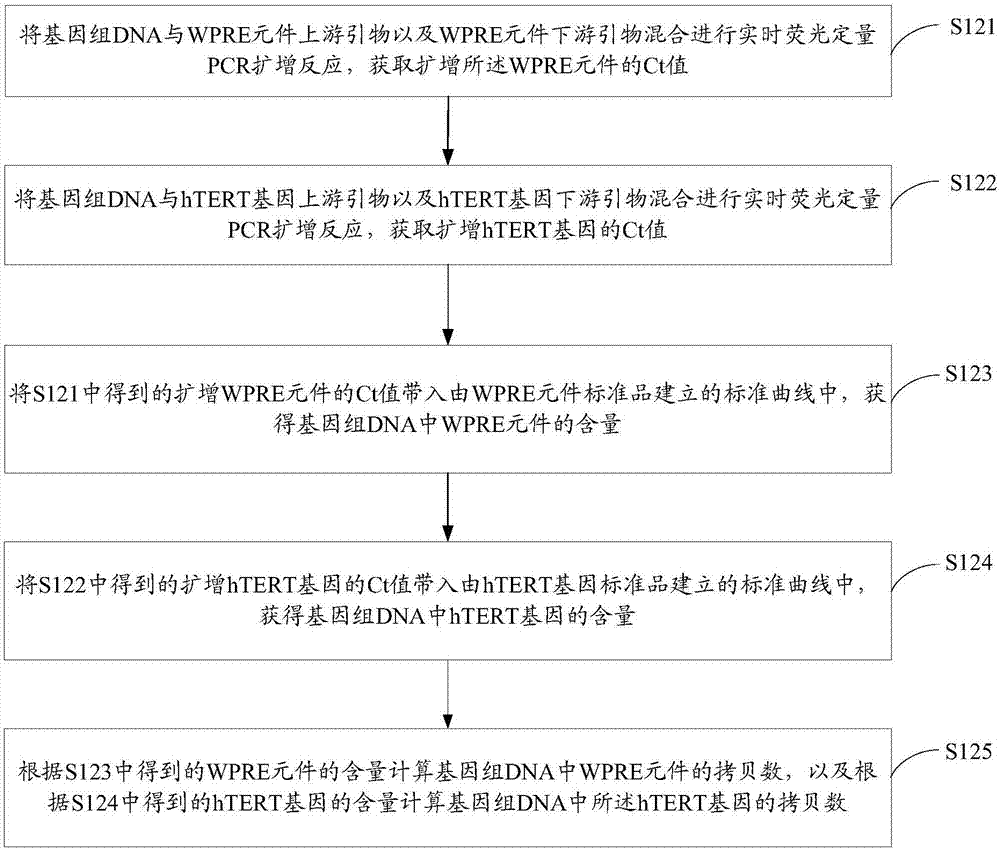
在一个实施方式中,请参阅图2,测定基因组dna中wpre元件的拷贝数以及htert基因的拷贝数的步骤包括以下操作s121~s125。
s121、将基因组dna与wpre元件上游引物以及wpre元件下游引物混合进行实时荧光定量pcr扩增反应,获取扩增所述wpre元件的ct值。
其中,ct值表示荧光信号到达设定的域值时所经历的循环数。
在一个实施方式中,提取dna后,加入wpre元件上游引物以及wpre元件下游引物,进行实时荧光定量pcr扩增反应,得到扩增曲线,根据扩增曲线获得扩增wpre元件的ct值。
具体地,用实时荧光定量pcr扩增反应扩增wpre元件时,反应条件为:95℃30秒,进入循环;95℃5秒,60℃30秒,终点读板,共40个循环。
在一个实施方式中,wpre元件上游引物的碱基序列seqidno.1所示,所述wpre元件下游引物的碱基序列seqidno.2所示。wpre元件上游引物和wpre元件下游引物无引物二聚体,且退火温度差距较小,能够特异性的扩增基因组dna中的wpre元件。
s122、将基因组dna与htert基因上游引物以及htert基因下游引物混合进行实时荧光定量pcr扩增反应,获取扩增htert基因的ct值。
在一个实施方式中,提取dna后,加入htert基因上游引物以及htert基因下游引物,进行实时荧光定量pcr扩增反应,得到扩增曲线,根据扩增曲线获得扩增htert基因的ct值。
具体地,用实时荧光定量pcr扩增反应扩增htert基因时,反应条件为:95℃30秒,进入循环;95℃5秒,60℃30秒,终点读板,共40个循环。
在一个实施方式中,htert基因上游引物的碱基序列seqidno.3所示,所述htert基因下游引物的碱基序列seqidno.4所示。htert基因上游引物和htert基因下游引物无引物二聚体,且退火温度差距较小,能够特异性的扩增基因组dna中的htert基因。
s123、将s121中得到的扩增wpre元件的ct值带入由wpre元件标准品建立的标准曲线中,获得基因组dna中wpre元件的含量。
wpre元件标准品建立的标准曲线中具有ct值与wpre元件含量的对应关系,根据扩增wpre元件的ct值即可定量计算出基因组dna中wpre元件的含量,即wpre元件的含量。
在一个实施方式中,由wpre元件标准品建立的标准曲线通过如下操作得到:将wpre元件标准品进行梯度稀释,获得梯度浓度的wpre元件标准品,将每一个浓度的wpre元件标准品分别与wpre元件上游引物以及wpre元件下游引物混合进行实时荧光定量pcr扩增反应,获取扩增每一个浓度的wpre元件标准品的ct值;及根据wpre元件标准品的ct值与浓度的对应关系建立wpre元件标准品的标准曲线。
具体地,将质量为1ng的wpre元件标准品以8为倍数进行梯度稀释,稀释7个梯度(最小含量稀释了2097152倍),获得总共8个浓度的wpre元件标准品。然后以每一个浓度的wpre元件标准品分别与wpre元件上游引物以及wpre元件下游引物混合进行实时荧光定量pcr扩增反应,获取扩增每一个浓度的wpre元件标准品的ct值并根据wpre元件标准品的ct值与浓度的对应关系建立wpre元件标准品的标准曲线。
在一个实施方式中,wpre元件标准品通过如下操作得到:将wpre元件模板与wpre元件模板的上游引物以及wpre元件模板的下游引物混合进行实时pcr扩增反应,收集pcr扩增反应的产物,得到wpre元件标准品,其中,wpre元件模板的上游引物的碱基序列seqidno.5所示,wpre元件模板的下游引物的碱基序列seqidno.6所示。
具体地,用pcr扩增反应扩增wpre元件模板获得wpre元件标准品时,反应条件为98℃10s,1个循环;98℃10s,55℃10s,72℃60s,30个循环;72℃5min,1个循环。
具体地,wpre元件模板的序列参照genbank数据库中登录号nc_004107.1。seqidno.5所示的wpre元件模板的上游引物,以及seqidno.6所示wpre元件模板的下游引物无引物二聚体,退火温度差距较小,且能够特异性的扩增wpre元件模板,从而获得wpre元件标准品。
上述wpre元件标准品的制备方法简单易行、经济实惠,从而获得大量的wpre元件标准品。
当然,在其他实施方式中,还可以通过直接碱基合成的方式获得wpre元件标准品。
s124、将s122中得到的扩增htert基因的ct值带入由htert基因标准品建立的标准曲线中,获得基因组dna中htert基因的含量。
htert基因标准品建立的标准曲线中具有ct值与htert基因含量的对应关系,根据扩增htert基因的ct值即可定量计算出基因组dna中htert基因的含量,即htert基因的含量。
在一个实施方式中,由htert基因标准品建立的标准曲线通过如下操作得到:将htert基因标准品进行梯度稀释,获得梯度浓度的htert基因标准品,将每一个浓度的htert基因标准品分别与htert基因上游引物以htert基因下游引物混合进行实时荧光定量pcr扩增反应,获取扩增每一个浓度的htert基因标准品的ct值;及根据htert基因标准品的ct值与浓度的对应关系建立htert基因标准品的标准曲线。
具体地,将质量为1ng的htert基因标准品以8为倍数进行梯度稀释,稀释7个梯度(最小含量稀释了2097152倍),获得总共8个浓度的wpre元件标准品。然后以每一个浓度的htert基因标准品分别与htert基因上游引物以及htert基因下游引物混合进行实时荧光定量pcr扩增反应,获取扩增每一个浓度的htert基因标准品的ct值并根据htert基因标准品的ct值与浓度的对应关系建立htert基因标准品的标准曲线。
在一个实施方式中,htert基因标准品通过如下操作得到:将htert基因模板与htert基因模板的上游引物以及htert基因模板的下游引物混合进行pcr扩增反应,收集pcr扩增反应的产物,得到htert基因标准品,其中,htert基因模板的上游引物的碱基序列seqidno.7所示,htert基因模板的下游引物的碱基序列seqidno.8所示。
具体地,用pcr扩增反应扩增htert基因模板时获得htert基因标准品时,反应条件为98℃10s,1个循环;98℃10s,55℃10s,72℃60s,30个循环;72℃5min,1个循环。
具体地,htert基因模板的序列参照genbank数据库中登录号ng_009265.1。seqidno.7所示的htert基因模板的上游引物,以及seqidno.8所示htert基因模板的下游引物无引物二聚体,退火温度差距较小,且能够特异性的扩增htert基因模板,从而获得htert基因标准品。
上述htert基因标准品的制备方法简单易行、经济实惠,从而获得大量的htert基因标准品。
当然,在其他实施方式中,还可以通过直接碱基合成的方式获得htert基因标准品。
s125、根据s123中得到的wpre元件的含量计算基因组dna中wpre元件的拷贝数,以及根据s124中得到的htert基因的含量计算基因组dna中所述htert基因的拷贝数。
具体地,wpre元件的拷贝数=(na×wpre元件的含量)/wpre元件的标准分子量。
具体地,htert基因的拷贝数=(na×htert基因的含量)/htert基因的标准分子量。
其中,na表示阿伏伽德罗常数,wpre元件的标准分子量=一对碱基的平均分子量×wpre元件标准品的碱基对数,htert基因的标准分子量=一对碱基的平均分子量×htert基因标准品的碱基对数。
具体地,na=6.0221×1023,一对碱基的平均分子量约为660g/mol。wpre元件标准品的碱基对数约为329,htert基因标准品的碱基对数约为1035。
根据wpre元件的含量与wpre元件的标准分子量之间的关系以及htert基因的含量与htert基因的标准分子量之间的关系准确定量计算出wpre元件的拷贝数和htert基因的拷贝数。
上述测定基因组dna中wpre元件的拷贝数以及htert基因的拷贝数的方法简单易行、定量准确,从而精确的计算获得基因组dna中wpre元件的拷贝数以及htert基因的拷贝数。
当然,在其他实施方式中,还可以通过特定的检测试剂盒检测基因组dna中wpre元件的拷贝数以及htert基因的拷贝数。
s130、根据s120中得到的wpre元件的拷贝数和htert基因的拷贝数计算平均每个靶细胞中重组慢病毒的颗粒数,其中,平均每个靶细胞中重组慢病毒的颗粒数=(wpre元件的拷贝数/htert基因的拷贝数)×2。
htert基因是靶细胞中一个稳定的单拷贝基因,htert基因的表达水平可作为内参。重组慢病毒感染靶细胞后,将包含wpre元件的整个外源基因表达框导入到靶细胞的基因组中,用wpre元件的拷贝数除以htert基因的拷贝数再乘以2(要乘以2是因为靶细胞是二倍体,存在两个等位基因)即可获得平均每个靶细胞中重组慢病毒的颗粒数,即单个基因组中导入重组慢病毒基因的拷贝水平。
s140、根据靶细胞的总数和s130中得到的平均每个靶细胞中重组慢病毒的颗粒数计算单位体积的待测样品中含重组慢病毒的颗粒数,得到待测样品中重组慢病毒的滴度。
在一个实施方式中,根据靶细胞的总数和平均每个靶细胞中重组慢病毒的颗粒数计算单位体积的待测样品中含重组慢病毒的颗粒数的步骤中:
单位体积的待测样品中含重组慢病毒的颗粒数=(靶细胞的总数×平均每个靶细胞中重组慢病毒的颗粒数)/待测样品的体积。
平均每个靶细胞中重组慢病毒的颗粒数相当于转导单位(tu)与靶细胞的总数相乘后,得到总的靶细胞中重组慢病毒的颗粒数,再除以待测样品的体积,获得单位体积的待测样品含重组慢病毒的颗粒数,即待测样品中重组慢病毒的滴度。
上述定量检测重组慢病毒的滴度的方法,发明人在标记基因的选择、测算方法等方面进行了大量的探索研究,预料不到地发现通过测定感染重组慢病毒后的靶细胞的基因组dna中wpre元件的拷贝数以及htert基因的拷贝数即可准确测算重组慢病毒的滴度。该方法操作简便,能够在不借助荧光标记物的条件下准确定量测定重组慢病毒的滴度。
以下为具体实施例部分。
实施例中采用试剂和仪器如非特别说明,均为本领域常规选择。实施例中未注明具体条件的实验方法,通常按照常规条件,例如文献、书本中所述的条件或者试剂盒生产厂家推荐的方法实现。
其中,基因组dna提取试剂盒和dna胶回收试剂盒(北京天根公司),primestarhs(premix)和sybrpremixextaq(大连宝生物公司),293t细胞(美国atcc),dmem培养基和10%胎牛血清(美国gibco公司)。
实施例1
制备wpre元件标准品和htert基因标准品
参照genbank数据库中登录号nc_004107.1合成wpre元件模板的序列,按照primestarhs(premix)试剂盒推荐的体系加入seqidno.5所示的wpre元件模板的上游引物,以及seqidno.6所示wpre元件模板的下游引物,进行pcr扩增反应,反应条件为98℃10s,1个循环;98℃10s,55℃10s,72℃60s,30个循环;72℃5min,1个循环。
参照genbank数据库中登录号ng_009265.1合成htert基因模板的序列,按照primestarhs(premix)试剂盒推荐的体系加入seqidno.7所示的wpre元件模板的上游引物,以及seqidno.8所示htert基因模板的下游引物,进行pcr扩增反应,反应条件为98℃10s,1个循环;98℃10s,55℃10s,72℃60s,30个循环;72℃5min,1个循环。
反应结束后,用1%琼脂糖凝胶电泳检测wpre元件pcr产物和htert基因pcr产物的生成情况,其结果如图3所示。m表示为marker,1表示htert基因的pcr产物,2表示wpre元件的pcr产物。可以看到,wpre元件的pcr产物的位置处于250bp~500bp之间,htert基因的pcr产物的位置处于1000bp附近,与预期(329bp和1035bp)相符。且两者产物条带单一,未见杂带和拖带,说明wpre元件和htert基因的pcr特异性扩增非常成功。
用dna胶回收试剂盒分别回收两种pcr产物,然后用thermonanodrop2000分光光度计测定回收产物的浓度,再将其稀释至0.5ng/μl,获得wpre元件标准品和htert基因标准品。
实施例2
建立wpre元件标准品的标准曲线和htert基因标准品的标准曲线
取2μl浓度为0.5ng/μl的wpre元件标准品(质量为1ng),以8为倍数进行梯度稀释,稀释7个梯度(最小含量稀释了2097152倍),获得总共8个含量的wpre元件标准品。以这些8个含量的wpre元件标准品为模板,分别加入seqidno:1所示的wpre元件上游引物和seqidno:2所示的wpre元件下游引物进行实时荧光定量pcr扩增反应,反应条件为:95℃30秒,进入循环;95℃5秒,60℃30秒,终点读板,共40个循环。得到如图4所示的扩增曲线和如图5所示的熔解曲线。根据图4所示的扩增曲线获取扩增每一个含量的wpre元件标准品的ct值。根据wpre元件标准品的ct值与含量的对应关系建立wpre元件标准品的标准曲线如图6所示。曲线方程为y=-3.5542x+9.6608,r2=0.9937,其中横坐标为log10为底wpre元件标准品的含量的对数,纵坐标为wpre元件标准品的ct值。
取2μl浓度为0.5ng/μl的htert基因标准品(质量为1ng),以8为倍数进行梯度稀释,稀释7个梯度(最小含量稀释了2097152倍),获得总共8个含量的htert基因标准品。以这些8个含量的htert基因标准品为模板,分别加入seqidno:3所示的htert基因标准品上游引物和seqidno:4所示的htert基因标准品下游引物进行实时荧光定量pcr扩增反应,反应条件为:95℃30秒,进入循环;95℃5秒,60℃30秒,终点读板,共40个循环。得到如图7所示的扩增曲线和如图8所示的熔解曲线。根据图7所示的扩增曲线获取扩增每一个含量的htert基因标准品的ct值。根据htert基因标准品的ct值与含量的对应关系建立htert基因标准品的标准曲线如图9所示。曲线方程为y=-3.5389x+10.694,r2=0.9964,其中横坐标为log10为底htert基因标准品的含量的对数,纵坐标为htert基因标准品的ct值。
从图4所示的wpre元件标准品的扩增曲线和图7所示的htert基因标准品的扩增曲线可以看到,不同含量梯度的wpre元件标准品和htert基因标准品的扩增曲线在扩增曲线图上分布较为均匀,说明pcr扩增线性程度较好。
从图5所示的wpre元件标准品熔解曲线和图8所示的htert基因标准品的熔解曲线可以看到,wpre元件和htert基因的扩增产物均只有一个单峰,说明pcr产物特异性很好。
此外,从图6所示的wpre元件标准品的标准曲线以及图9所示的htert基因标准品的标准曲线可以看到,两者的标准曲线的r2均大于0.99,再次说明pcr的线性扩增程度好。
进一步地,计算荧光定量pcr扩增反应扩增wpre元件引物的效率,wpre元件引物的扩增效率=10(-1/slope)-1×100%=91.14%,htert基因引物的扩增效率=10(-1/slope)-1×100%=91.68%,其中,slope表示标准曲线的斜率。两者均落在80%~120%之间。说明wpre元件和htert基因的定量pcr引物能很好地用于后续的重组慢病毒滴度测定的研究中,seqidno.1所示的wpre元件上游引物以及seqidno.2所示的wpre元件下游引物能够特异性的扩增wpre元件。seqidno.3所示的htert基因上游引物以及seqidno.4所示的htert基因下游引物能够特异性的扩增htert基因。
实施例3
重组慢病毒滴度的定量测定
取2.5×106个293t细胞接种于10cm培养皿中,培养18h后弃除培养基,取3ml待测样品(即含重组慢病毒的病毒悬液)与6ml的dmem完全培养基,加入终浓度为4μg/ml的polybrene后混匀,一并将其加入到培养皿。24h后再次将含重组慢病毒的培养基替换为dmem完全培养基,继续培养72h后收集293t细胞,用基因组dna提取试剂盒提取其基因组dna。
按照primestarhs(premix)试剂盒推荐的体系以基因组dna为扩增模板,加入seqidno.1所示的wpre元件上游引物以及seqidno.2所示的wpre元件下游引物进行实时荧光定量pcr扩增反应,反应条件为:95℃30秒,进入循环;95℃5秒,60℃30秒,终点读板,共40个循环。通过荧光定量pcr仪记录基因组dna的扩增情况,记录所得的ct值,本实施例中扩增wpre元件的ct值为29.17。
按照primestarhs(premix)试剂盒推荐的体系以基因组dna为扩增模板,加入seqidno.3所示的htert基因上游引物以及seqidno.4所示的htert基因下游引物进行实时荧光定量pcr扩增反应,反应条件为:95℃30秒,进入循环;95℃5秒,60℃30秒,终点读板,共40个循环。通过荧光定量pcr仪记录基因组dna的扩增情况,记录所得的ct值,本实施例中扩增htert基因的ct值为31.21。
将扩增wpre元件的ct值带入图6所示的标准曲线中,计算得到基因组dna中wpre元件的含量=3.24×10-6ng。
接着计算wpre元件的拷贝数(copiesofwpre)=(na×wpre元件的含量)/wpre元件的标准分子量=6.0221×1023×3.24×10-6ng×10-9÷(660g/mol×329)=8.99×103。
将扩增htert基因的ct值带入图9所示的标准曲线中,计算得到基因组dna中htert基因的含量=1.59×10-6ng。
接着计算htert基因的拷贝数(copiesofhtert)=(na×htert基因的含量)/htert基因的标准分子量=6.0221×1023×1.59×10-6ng×10-9÷(660g/mol×1035)=1.40×103。
计算平均每个293t细胞中重组慢病毒的颗粒数(particlesof293t)=(wpre元件的拷贝数/htert基因的拷贝数)×2=(8.99×103÷1.40×103)×2=12.84。
然后计算单位体积的待测样品中含重组慢病毒的颗粒数(即待测样品中重组慢病毒滴度,titer,tu/ml)=(靶细胞的总数×平均每个靶细胞中重组慢病毒的颗粒数)/待测样品的体积=(2.5×106×12.84)÷3=1.07×107。
上述定量检测重组慢病毒的滴度的方法操作简便,能够在不借助荧光标记物的条件下准确定量测定重组慢病毒的滴度。
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
sequencelisting
<110>深圳精准医疗科技有限公司
<120>定量检测重组慢病毒的滴度的方法
<160>8
<170>patentinversion3.3
<210>1
<211>20
<212>dna
<213>人工序列
<400>1
ggcactgacaattccgtggt20
<210>2
<211>20
<212>dna
<213>人工序列
<400>2
agggacgtagcagaaggacg20
<210>3
<211>19
<212>dna
<213>人工序列
<400>3
aaatgcggcccctgtttct19
<210>4
<211>19
<212>dna
<213>人工序列
<400>4
cagtgcgtcttgaggagca19
<210>5
<211>15
<212>dna
<213>人工序列
<400>5
tgctgacgcaacccc15
<210>6
<211>15
<212>dna
<213>人工序列
<400>6
ggcgaaggcgaagac15
<210>7
<211>19
<212>dna
<213>人工序列
<400>7
accaagcacttcctctact19
<210>8
<211>17
<212>dna
<213>人工序列
<400>8
tgcaaaacccttacctc17
- 还没有人留言评论。精彩留言会获得点赞!