一种高对映选择性C‑5位α‑立体中心4‑硝基异恶唑醇类化合物、其制备方法及应用与流程
本发明属于药物合成领域,具体涉及一种高对映选择性c-5位α-立体中心4-硝基异恶唑醇类化合物、其制备方法及应用。
背景技术:
异恶唑是一类非常重要的、广泛用于有机合成具有杂环结构的化合物。这类化合物在降低人类血糖、消除人类的痛、抵抗人类的炎症、杀死有害细菌和控制和减小艾滋病毒的危害等方面对人类有很大的促进作用。此外,一些异恶唑衍生物也表现出农药化学效用,具有抑制杂草和土壤细菌生长的效能,因此,它在农药和杀虫剂领域也有着广泛的应用前景。异恶唑环结构也存在于某些天然产物,如类固醇类药物达那唑含有异恶唑环。鹅膏蕈氨酸,以及一些药物,包括cox-2抑制剂伐地考昔(商品名bextra)。一些耐β-内酰胺酶的抗生素中含有异恶唑环,例如氯唑西林,双氯青霉素,氟氯西林。
目前,有机不对称催化合成手性4-硝基异恶唑c-5位具有α-立体中心的异恶唑醇结构类衍生物的方法还没有被报道。因此,发展4-硝基异恶唑和多聚甲醛在氨基酸衍生的二肽-胍相转移催化剂作用下的不对称催化羟醛缩合反应,得到一类高对映选择性c-5位具有α-立体中心的异恶唑醇类化合物就变得更加重要。氨基酸衍生的二肽-胍相转移催化剂
吡唑在c-4位置上含有不同类型的芳基、在c-5位置上有不同的功能化官能团取代的具有α立体中心结构的化合物是一种很重要的杀菌剂。非手性的吡唑化合物
此类高对映选择性c-5位α-立体中心4-硝基异恶唑醇类化合物制备方法的不对称发展能够快速实现一系列手性异恶唑以及吡唑类化合物或者药物的高速发展。
技术实现要素:
本发明的目的在于提供一种高对映选择性c-5位α-立体中心4-硝基异恶唑醇类化合物、其制备方法及应用。
一种高对映选择性c-5位α-立体中心4-硝基异恶唑醇类化合物,其结构式如下:
上述高对映选择性c-5位α-立体中心4-硝基异恶唑醇类化合物,合成路线如下:
合成步骤为:合成步骤为:将化合物1、催化剂a、乙酸钠和
所述催化剂a的合成路线如下:
具体合成步骤如下:
(1)将化合物c1加入体积比为4︰1的水和thf的混合溶剂中,加入na2co3、boc2o,室温反应至结束,反应结束后在冰浴下调ph为2,加入乙酸乙酯萃取,有机相用na2so4干燥,旋干,柱层析分离即得化合物c2;
(2)在氮气氛围和-78℃下,加入钠块,通入氨气,形成液态氨基钠,加入化合物c2的thf溶液,并在-78℃至反应完全,升至室温,加入nh4cl固体进行猝灭,室温搅拌直至钠块消耗完全,抽滤,取滤液旋干即得化合物c3;
(3)将化合物c4、二氯甲烷加入圆底烧瓶中,依次加入edci、dmap和化合物c3,室温反应完全,柱层析分离即得化合物c5;
(4)在氮气氛围和-78℃下,加入钠块,通入氨气,形成液态氨基钠,加入化合物c5的thf溶液,并在-78℃至反应完全,升至室温,加入nh4cl固体进行猝灭,室温搅拌直至钠块消耗完全,抽滤,取滤液旋干即得化合物c6;
(5)将化合物c6溶于二氯甲烷中,加入3,5-二氯异氰酸苯酯,室温反应完全,柱层析分离即得化合物c7;
(6)将化合物c7溶于甲醇中,滴加accl,室温反应至完全,旋干,即得化合物c8;
(7)在氮气氛围下,加入化合物c8,在0℃依次加入et3n、化合物c9的二氯甲烷溶液,室温搅拌至反应完全,柱层析分离,即得催化剂a。
较好地,所述步骤(1)中,c1、na2co3和boc2o的摩尔比为1︰1︰1.2;所述步骤(2)中,化合物c2和钠块的摩尔比为50︰1;所述步骤(3)中,化合物c4、edci、dmap、化合物c3的摩尔比为1.2︰1.3︰0.2︰1;所述步骤(4)中,化合物c5和钠块的摩尔比为50︰1;所述步骤(5)中,化合物c6与3,5-二氯异氰酸苯酯的摩尔比为1.2︰1;所述步骤(6)中,化合物c7与accl的摩尔比为10︰1;所述步骤(7)中,化合物c8、et3n和化合物c9的摩尔比为1︰2︰1.5。
进一步地,化合物1、化合物2、催化剂a和acona的摩尔比为1.0:3.0:0.1:1.0。
每0.1mmol化合物1添加50.0mg
上述高对映选择性c-5位α-立体中心4-硝基异恶唑醇类化合物作为起始物在合成吡唑类杀真菌剂药物中例如(cas:1845899-33-3)的应用。
本发明可以高效制备高对映选择性c-5位α-立体中心4-硝基异恶唑醇类化合物,本发明的优势在于:反应成本低,效率高,反应路径合理,后处理简单,得到高光学纯度化合物和高立体选择性化合物。
附图说明
图1是催化剂a的1hnmr谱图;
图2是化合物3a的1hnmr谱图;
图3是化合物3b的1hnmr谱图;
图4是化合物3c的1hnmr谱图;
图5是化合物3d的1hnmr谱图;
图6是化合物7的1hnmr谱图。
具体实施方式
以下结合具体实施例对本发明的技术方案作进一步详细说明,但本发明的保护范围并不局限于此。
仪器与主要化学试剂
brukerav-300型核磁共振仪(德国);agilent1200高效液相色谱仪(美国)。
本发明实施过程中所用的原料、溶剂均为商业途径购进。
下述各实施例中的催化剂a通过下述方法合成:
步骤1:将白色固体l-叔亮氨酸衍生的手性二胺化合物c1(具体合成步骤参考ye,w.;leow,d.;tan,c.h.tetrahedronletters.2006,47(6),1007-1010)(589.4mg,2.18mmol,1.0equiv)转移至盛有水(20ml)和thf(5ml)的100ml烧瓶内,依次加入na2co3(231.1mg,2.18mmol1.0equiv)、boc2o(1.2equiv),室温搅拌12小时。反应结束,在冰浴下,将反应液2m盐酸调至ph=2,加乙酸乙酯萃取(20ml×3),有机相用na2so4干燥,旋干。干法上柱(洗脱剂:石油醚和乙酸乙酯体积比从20:1到10:1),得纯品白色固体c2(产率为92%)。
步骤2:取100ml两口瓶,烤瓶子,抽真空,换氮气。-78℃加入切好的钠块儿(50.0equiv),缓慢通入氨气,形成液态氨基钠,将上一步产物c2(741.0g,2.0mmol,1.0equiv)溶于thf,用注射器缓慢注入液态氨基钠中,在-78℃下,反应4小时。升至室温,缓慢加入nh4cl固体进行猝灭,室温搅拌过夜直至钠块儿消耗完全。抽滤,取滤液,旋干得到白色固体c3(产率为95%),直接用于下一步。
步骤3:取100ml圆底烧瓶,加入已制备好的ts保护的l-叔亮氨酸c4(650.7mg,2.28mmol,1.2equiv),加25mldcm做溶剂,依次加入edci(1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐,473.5mg,2.47mmol,1.3equiv)、dmap(4-二甲氨基吡啶,46.2mg,0.38mmol,0.2equiv),最后将上一步得到白色固体c3(411.0mg,1.9mmol,1.0equiv)加入圆底烧瓶内。室温搅拌12小时,加水,乙酸乙酯萃取,有机相用na2so4干燥,旋干溶剂,柱层析分离(洗脱剂:乙酸乙酯和正己烷体积比1:10),得纯品白色固体c5(产率为82%)。
步骤4:取100ml两口瓶,烤瓶子,抽真空,换氮气。加入切好的钠块儿(50.0equiv),放入-78℃,缓慢通入氨气,形成液态氨基钠,将产物c5(725.6mg,1.5mmol,1.0equiv.)溶于thf,用注射器缓慢加入液态氨基钠,在-78℃下,反应4小时。升至室温,缓慢加入nh4cl固体进行猝灭,室温搅拌过夜直至钠块儿消耗完全。抽滤,取滤液,旋干得到白色固体c6(产率为95%),直接用于下一步。
步骤5:将上一步白色固体c6(428.4mg,1.3mmol,1.0equiv)转移至100ml圆底烧瓶,加5mldcm做溶剂,加入3,5-二氯异氰酸苯酯(293.4mg,1.56mmol,1.2equiv),室温搅拌2小时,旋干溶剂,柱层析分离(洗脱剂:二氯甲烷和甲醇体积比从100:1到30:1),得纯品白色固体c7(产率为78%)。
步骤6:将上一步白色固体c7(517.5mg,1.0mmol,1.0equiv)转移至100ml圆底烧瓶,加5mlmeoh做溶剂,向反应液中缓慢滴加accl(乙酰氯,0.707ml,10.0mmol,10.0equiv),室温搅拌20小时,直至原料反应完全。旋干,得白色固体c8。真空抽干,直接用于下一步(产率>99%)。
步骤7:取100ml两口瓶,烤瓶子,抽真空,换氮气。在氮气环境,将上一步白色固体c8(417.4mg,1.0mmol,1.0equiv)加入到两口瓶,在冰浴下(0℃),用注射器将et3n(tea,0.279ml,2.0mmol,2.0equiv)缓慢加入,最后将化合物c9(具体合成步骤参考ma,t.;fu,x.;huang,k.w.;tan,c.h.j.am.chem.soc.2011,133(9),2828-2831)(710.1mg,1.5mmol,1.5equiv)溶于5mldcm,缓慢加入到上述反应体系中。室温搅拌直至原料反应完全(约2h)。旋干溶剂,柱层析分离(洗脱剂,二氯甲烷和甲醇体积比从100:1到50:1),得纯品白色固体即为催化剂a(产率为67%)。
催化剂a,mp142.3-144.5℃;
实施例1:
化合物3a的结构式如下:
产物名称:(r)-2-(3-甲基-4-硝基异恶唑-5)-丙烷-1醇。
化合物3a的合成路线如下:
化合物3a的合成步骤如下:
(1)将3-甲基-5-乙基-4-硝基异恶唑1a(0.1mmol,1.0equiv.)、催化剂a(0.01mmol,0.1equiv.)、乙酸钠(0.1mmol,1.0equiv)和
(2)加入多聚甲醛2a(0.3mmol,3.0equiv.),室温继续反应直至通过薄层板(tlc)监测原料1a完全消失(约70h);
(3)旋干溶剂,柱层析分离(洗脱剂:石油醚和乙酸乙酯体积比从10:1到3:1),得到无色油状液体3a(85%产率)。
无色油状液体,97%ee,
化合物的ee值用高效液相色谱法(hplc)分析得到,chiralpakic(4.6mm×250mmi.d.),正己烷/异丙醇=80/20(v/v),流速1.0ml/min,25℃,254nm,tr=5.8min(小峰)和7.4min(大峰)。
实施例2:
产物名称:产物名称:(r)-2-(3-乙基-4-硝基异恶唑-5)-丙烷-1醇。
步骤(1)中,用3,5-乙基-4-硝基异恶唑1b替换3-甲基,5-乙基,4-硝基异恶唑1a,其它实验步骤及提纯方式参照实施例1进行;淡黄色油状物,97%ee,86%yield,
化合物的ee值用高效液相色谱法(hplc)分析得到,chiralpakie(4.6mm×250mmi.d.),正己烷/异丙醇=80/20(v/v),流速1.0ml/min,25℃,254nm,tr=7.8min(小峰)和8.3min(大峰)。
实施例3:
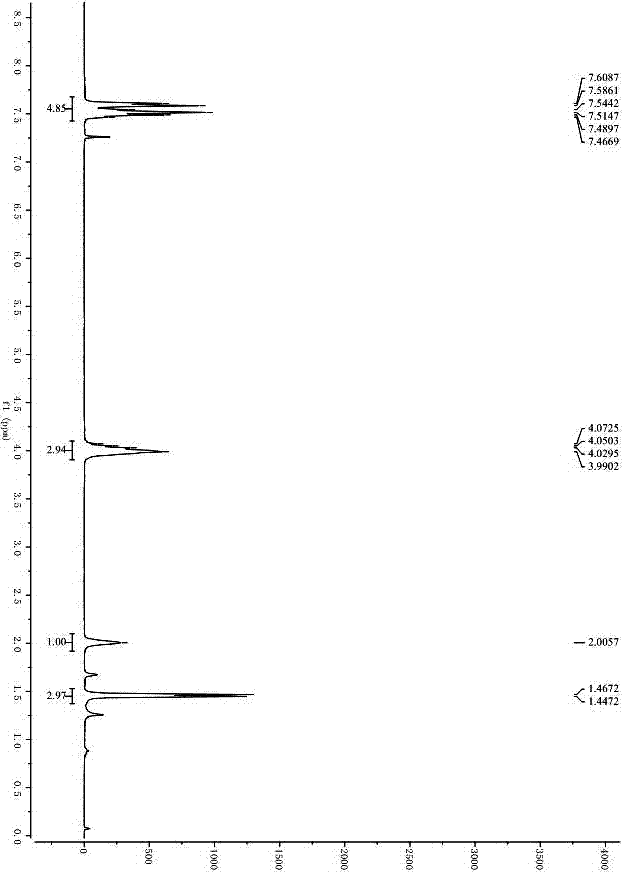
步骤(1)中,用3-苯基-5-乙基-4-硝基异恶唑1c代替3-甲基,5-乙基,4-硝基异恶唑1a,其它实验步骤及提纯方式参照实施例1步骤进行;淡黄色油状物,85%ee,95%yield,1hnmr(300mhz,cdcl3)δ7.47-7.61(m,5h),4.00-4.07(m,3h),2.01(s,1h),1.46(d,j=6.0hz,3h),具体如图4所示;13cnmr(75mhz,cdcl3)δ176.6,157.9,130.7,129.7,129.2,128.5,125.7,64.9,36.3,13.9;hrms(esi)m/z249.0877(m+h+),calc.forc12h13n2o4249.0875。
化合物的ee值用高效液相色谱法(hplc)分析得到,chiralpakie(4.6mm×250mmi.d.),正己烷/异丙醇=80/20(v/v),流速1.0ml/min,25℃,254nm,tr=7.6min(小峰)和9.2min(大峰)。
实施例4:
产物名称:(r)-2-(3-噻吩基-4-硝基异恶唑-5)-庚烷-1-醇。
步骤(1)中,用3-噻吩基-5-丁基-4-硝基异恶唑1d代替3-甲基-5-乙基-4-硝基异恶唑1a,其它实验步骤及提纯方式参照实施例1步骤进行;淡黄色油状物,92%ee,94%yield,
化合物的ee值用高效液相色谱法(hplc)分析得到,chiralpakid-3(4.6mm×250mmi.d.),正己烷/异丙醇=80/20(v/v),流速1.0ml/min,25℃,254nm,tr=6.9min(小峰)和8.2min(大峰)。
采用本发明的方法合成了一种高对映选择性c-5位α-立体中心4-硝基异恶唑醇类化合物,此类方法目前尚未有报道。该方法具有如下优势:本发明可以通用,成本低,高立体选择性和高光学活性,反应路径合理,高效制备该类型的化合物。
应用实验
用不对称催化羟醛缩合反应产物得到高对映选择性c-5位具有α-立体中心的异恶唑醇类化合物通过一系列的合成反应能够得到手性杀真菌剂
化合物7的合成步骤如下:
(a)将化合物3(52.1mg,0.28mmol)溶解于乙醚(1.0ml)中,室温搅拌,加入水合肼(0.0143ml,0.294mmol),继续室温搅拌18小时【通过薄层板(tlc)监测原料3完全消失】,旋干溶剂,柱层析分离(洗脱剂:石油醚和乙酸乙酯体积比从20:1到1:1),得到化合物4(无色油状液体,72%产率);
(b)将化合物4(37.04mg,0.2mmol)溶解于四氢呋喃(2.0ml)中,0°搅拌下加入氢化钠(24.0mg,1.0mmol),继续搅拌15分钟,然后用注射器加入碘甲烷(0.062ml,1.0mmol)。放置室温,继续搅拌12小时【通过薄层板(tlc)监测原料4完全消失】,旋干溶剂,柱层析分离(洗脱剂:石油醚和乙酸乙酯体积比从30:1到5:1),得到化合物5(无色油状液体,88%产率);
(c)将化合物5(27.7mg,0.13mmol)、六水合氯化镍(30.85mg,0.13mmol)溶解于甲醇(2.0ml)中-40℃搅拌10分钟,加入硼氢化钠(14.8mg,0.39mmol),继续-40℃搅拌3小时【通过薄层板(tlc)监测原料5完全消失】,过滤反应,除去滤渣,滤液中加入水,乙酸乙酯萃取,收取乙酸乙酯有机相层,用无水硫酸钠干燥除水。旋干溶剂,柱层析分离(洗脱剂:二氯甲烷和甲醇体积比从100:1到30:1),得到化合物6(无色油状液体,87%产率);
(d)氩气氛围下,将2,6-二甲基溴苯(21.78ml,0.164mmol),叔丁醇钠(20.95mg,0.218mmol),三(二亚芐基丙酮)二钯(9.98mg,0.0109mmol)和1,1'-联萘-2,2'-双二苯膦(13.57mg,0.0218mmol)依次加入圆底瓶反应中,化合物6(20.0mg,0.109mmol)溶解于甲苯(2.0ml)中并将化合物的甲苯溶液加入圆底瓶中。反应放置于90℃油浴中加热17小时【通过薄层板(tlc)监测原料6完全消失】,冷却反应,过滤,用四氢呋喃洗,收集滤液,旋干溶剂,柱层析分离(洗脱剂:二氯甲烷和甲醇体积比从100:1到30:1),得到化合物7(红色油状液体,71%产率)。
94%ee,
化合物的ee值用高效液相色谱法(hplc)分析得到,chiralpakie(4.6mm×250mmi.d.),正己烷/异丙醇=90/10(v/v),流速1.0ml/min,25℃,254nm,tr=7.5min(大峰)和8.9min(小峰)。
上述实验结果表明:高对映选择性3a通过4步合成反应得到高对映选择性杀真菌剂7(s)-n-(2,6-二甲基苯基)-5-(2-(1-甲氧基丙烷))-1,3-二甲基-1h-吡唑-4-胺(cas:1845899-33-3),反应路径合理。
同时,用合成的高对映选择性产物3起始原料合成一系列类似杀真菌剂(cas:1845899-33-3)的化合物,为杀真菌剂药物筛选提供帮助。
最后应说明的是:以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。
- 还没有人留言评论。精彩留言会获得点赞!