一种合成2,2‑二氟己酸乙酯的方法与流程
本发明涉及一种无金属催化合成烯烃的氢化二氟乙酸乙酯化产物的方法,尤其是一种合成2,2-二氟己酸乙酯的方法,属于有机化合物的合成及分离技术领域。
背景技术:
含氟官能团由于其独特的生理医药特性,可以改变药物的亲酯和药物代谢等特性,而被广泛应用于医药的合成设计中。其中最便捷的引入含氟官能团的办法是烯烃与氟试剂直接反应得到。例如(s)-2,2-二氟-n-癸酰基-l-高丝氨酸内酯类化合物革兰氏阴性菌群体感应系统中最重要的一类信号分子,并调控许多生理特性的表达。2,2-二氟己酸乙酯是鲁比前列酮合成的重要前驱体,而鲁比前列酮是氯离子通道激活剂,可选择性活化位于胃肠道上皮尖端管腔细胞膜上的2型氯离子通道,增加肠液的分泌和肠道的运动性,从而增加排便,减轻慢性特发性便秘的症状,且不改变血浆中钠和钾的浓度。
目前已经报道的合成2,2-二氟己酸乙酯类衍生物常采用以下几种方法:(1)由dinoalberico等人报道的在0℃下将氟化试剂dast滴加入2-羰基己酸乙酯二氯甲烷溶液中,再恢复室温反应4小时,得到2,2-二氟己酸乙酯。这种方法原料价格较高,反应原料受限。(美国专利us20100056808a1)。
(2)采用金属zn/nicl2催化合成2,2-二氟己酸乙酯。这种方法最早由杨振宇(j.org.chem.,1992,57,5144.)等人报道的通过碘二氟乙酸乙酯与1-丁烯在zn/nicl2催化下合成得到,但是碘二氟乙酸乙酯价格昂贵,后来张巍等人将这种方法改进用溴二氟乙酸乙酯替代碘二氟乙酸乙酯,但此种方法往往存在金属残留问题而且操作麻烦。
(3)chunghyeonyu(chem.commun.,2014,50,12884.)等人报道了由烯烃与溴代二氟乙酸乙酯在光催化剂fac-ir(ppy)3催化下以tmeda/dbu作为还原剂及碱得到十二烷基二氟乙酸乙酯。这种方法使用的光催化剂价格昂贵,且常常伴随各种卤化产物产生。
技术实现要素:
本发明的目的在于解决现有技术的不足,提供一种合成2,2-二氟己酸乙酯的方法,其无需用到金属催化剂,并且原料易得、成本低,反应条件温和。
技术方案
本发明的合成路线如下:
改变原料,利用本发明的合成方法还可合成重要的化工原料c14h18f2o3和c14h18f2o2,合成路线如下:
本发明的具体技术方案为:
一种合成2,2-二氟己酸乙酯的方法,包括如下步骤:
(1)在玻璃反应容器中,加入二苯二硫醚及2,6-二甲基-1,4-二氢-3,5-吡啶二羧酸二乙酯,用氮气置换玻璃反应容器中的空气后,加入丁烯和溴二氟乙酸乙酯,再加入溶剂;
(2)将玻璃反应容器置于蓝光led灯或者白光节能灯下照射12-48h,然后除去产物中的溶剂,加入石油醚将产物溶解,采用以300-400目硅胶作为固定相的柱层析技术,湿法上样,以石油醚为洗脱剂进行洗脱,gc-ms检测产物带,并将收集的层析液旋干,即得2,2-二氟己酸乙酯。
步骤(1)中,丁烯和溴二氟乙酸乙酯的摩尔比为3:1。
步骤(1)中,二苯二硫醚与溴二氟乙酸乙酯的摩尔比为0.05:1,2,6-二甲基-1,4-二氢-3,5-吡啶二羧酸二乙酯与溴二氟乙酸乙酯的摩尔比为2:1。
步骤(1)中,所述溶剂为二氯甲烷。
步骤(2)中,除去产物中溶剂的方法是采用旋转蒸发器旋干溶剂。
步骤(2)中,洗脱剂石油醚的沸程为30-60℃。
本发明的优点:本发明的合成方法操作简单,对溶剂质量要求低,可以应于多种烯烃,原料易得,成本低,反应条件温和,并且采用无金属催化,避免了金属残留。
附图说明
图1为实施例1制得的产物在氘代氯仿中的1hnmr图谱;
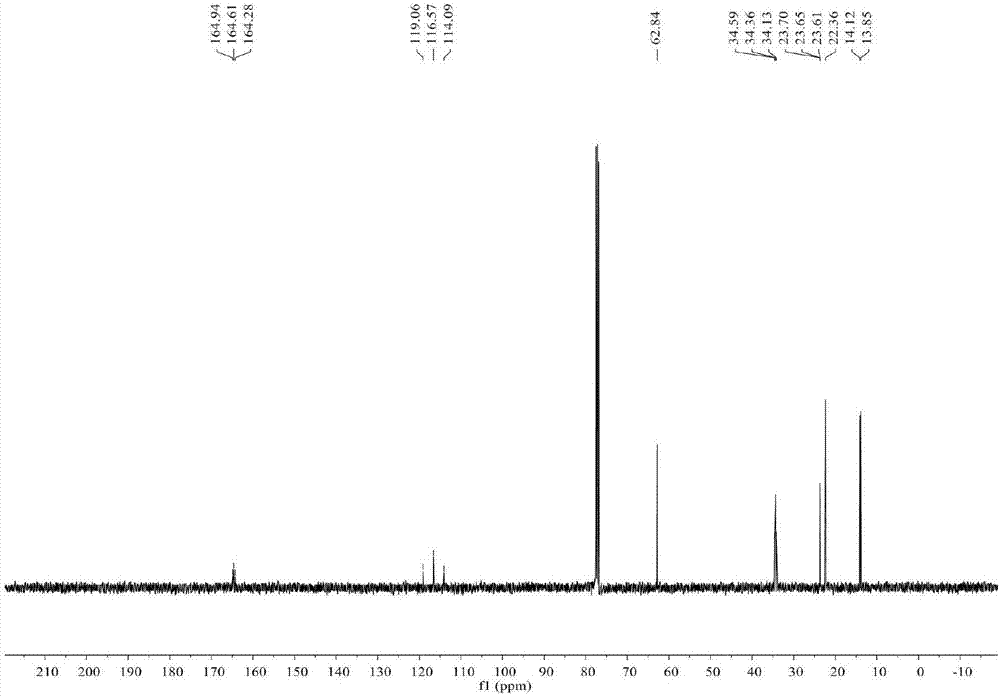
图2为实施例1制得的产物在氘代氯仿中的13cnmr图谱;
图3为实施例1制得的产物在氘代氯仿中的19fnmr图谱。
具体实施方式
下面结合附图和具体实施例对本发明作进一步说明。
实施例1
在100ml反应试管中,加入二苯二硫醚(109mg,0.5mmol)及2,6-二甲基-1,4-二氢-3,5-吡啶二羧酸二乙酯(5.06g,20mmol),在n2线上用氮气置换管中空气后换后,在氮气条件下丁烯正己烷溶液(10%正己烷溶液约15ml,30mmol)和溴二氟乙酸乙酯(2.03g10mmol),再加入30ml二氯甲烷。将反应容易置于蓝光led灯或者白光节能灯下照射48小时后用旋转蒸发器旋干溶剂,加入石油醚(沸程30-60℃)溶解。采用以300-400目硅胶作为固定相的柱层析技术,湿法上样,以石油醚(沸程30-60℃)为洗脱剂进行洗脱,gc-ms检测产物带,并将收集的层析液旋干,即可得到产物2,2-二氟己酸乙酯(780mg,产率43%)。
产物在氘代氯仿中的1hnmr图谱见图1,在氘代氯仿中的13cnmr图谱见图2,在氘代氯仿中的19fnmr图谱见图3。
实施例2
合成c14h18f2o3:
在10ml透明反应试管中,加入二苯二硫醚(2.2mg,0.01mmol)及2,6-二甲基-1,4-二氢-3,5-吡啶二羧酸二乙酯(101.2mg,0.4mmol),放置于手套箱中,用氮气置换后,用聚四氟乙烯内村垫片封住瓶口后,从手套箱中拿出,用微量进样器加入对甲氧基苯丙烯(29.6mg,0.2mmol)和溴二氟乙酸乙酯(81.2mg,0.4mmol),再加入2ml乙腈。将反应容易置于蓝光led灯或者白光节能灯下照射12-48小时后用旋转蒸发器旋干溶剂,加入正己烷溶解。采用以300-400目硅胶作为固定相的柱层析技术,湿法上样,以正己烷为洗脱剂进行洗脱,gc-ms检测洗脱液,并将收集的层析液旋干,即可得到产物1(24.0mg,产率44%)。
实施例3
合成c14h18f2o2:
在10ml透明反应试管中,加入二苯二硫醚(2.2mg,0.01mmol)及2,6-二甲基-1,4-二氢-3,5-吡啶二羧酸二乙酯(101.2mg,0.4mmol),放置于手套箱中,用氮气置换后,用聚四氟乙烯内村垫片封住瓶口后,从手套箱中拿出,用微量进样器加入苯丁烯(29.6mg,0.2mmol)和溴二氟乙酸乙酯(81.2mg,0.4mmol),再加入2ml乙腈。将反应容易置于蓝光led灯或者白光节能灯下照射12-48小时后用旋转蒸发器旋干溶剂,加入正己烷溶解。采用以300-400目硅胶作为固定相的柱层析技术,湿法上样,以正己烷为洗脱剂进行洗脱,gc-ms检测产物带,并将收集的层析液旋干,即可得到产物2(33.3mg,产率65%)。
- 还没有人留言评论。精彩留言会获得点赞!