一种Reltecimod的液相制备方法与流程
本发明涉及医药领域,具体涉及一种reltecimod的制备方法。
背景技术:
reltecimod是atoxbio公司正在开发的用于治疗由嗜肉细菌引起的坏死性软组织重度感染药物。reltecimod是由天然氨基酸组成的八肽,化学名为2-((5s,11s,14s,17s)-5-(1-((s)-2-amino-3-hydroxypropanoyl)pyrrolidine-2-carboxamido)-17-(4-hydroxybenzyl)-8-isobutyl-11-isopropyl-14-methyl-6,9,12,15-tetraoxo-2-thia-7,10,13,16-tetraazaoctadecanamido)succinicacid,分子式为c40h62n8o13s,分子量为895.03,其结构如下:
嗜肉细菌传播速度很快,经常引起重大组织损坏和全身性疾病,从而导致多种器官功能障碍、衰竭和死亡。除抗生素外,现今并没有针对嗜肉细菌的标准治疗方法,reltecimod将提供一种具有显著意义的非抗生素治疗方案。
现有公开文献中没有关于reltecimod的合成方法的报导,因此提供一种可工业化生产的工艺显得十分必要。
肽类药物的合成一般采用固相合成方法,该方法需要采用固体载体并通过过量投料保证反应完全,成本较高,经济价值不大。
技术实现要素:
为了解决上述技术问题,本发明一个方面提供了一种reltecimod的制备方法,其包括如下步骤:
步骤1),合成三个全保护的多肽片段,r1-ser(r4)-pro-oh,r2-met-leu-val-oh和r3-ala-tyr(r5)-asp(r6)-oh;
步骤2)偶联r2-met-leu-val-oh和r3-ala-tyr(r5)-asp(r6)-oh,得到r2-met-leu-val-ala-tyr(r5)-asp(r6)-oh;
步骤3)偶联r1-ser(r4)-pro和r2-met-leu-val-ala-tyr(r5)-asp(r6)-oh,得到r1-ser(r4)-pro-met-leu-val-ala-tyr(r5)-asp(r6)-oh;
步骤4)脱除保护基,得到粗品;
步骤5)纯化后得到reltecimod纯品;
其中,步骤r1、r2和r3均为氨基保护基,r4为丝氨酸侧链保护基,r5为酪氨酸侧链保护基,r6为天冬氨酸侧链保护基。
在本发明的技术方案中,合成r1-ser(r4)-pro-oh的方法为,将r1-ser(r4)-oh进行羧基活化,然后与nh2-pro-oh在液相中进行偶联。
在本发明的技术方案中,合成r2-met-leu-val-oh的方法为,将r7-leu-oh进行羧基活化,然后与nh2-val-oh进行液相偶联,得到r7-leu-val-oh,脱除氨基保护基r7,得到nh2-leu-val-oh;将r2-met-oh进行羧基活化,然后与nh2-leu-val-oh进行液相偶联,得到r2-met-leu-val-oh。
在本发明的技术方案中,合成r3-ala-tyr(r5)-asp(r6)-oh的方法为将r8-tyr(r5)oh进行羧基活化,然后与nh2-asp(r6)-oh进行液相偶联,得到r8-tyr(r5)-asp(r6)-oh,脱除氨基保护基r8,得到nh2-tyr(r5)-asp(r6)-oh;将r3-ala-oh进行羧基活化,然后与nh2-tyr(r5)-asp(r6)-oh进行液相偶联,得到r3-ala-tyr(r5)-asp(r6)-oh。
在本发明的技术方案中,合成r2-met-leu-val-ala-tyr(r5)-asp(r6)-oh的方法为r3-ala-tyr(r5)-asp(r6)-oh脱除氨基保护基r3,得到nh2-ala-tyr(r5)-asp(r6)-oh;将r2-met-leu-val-oh进行羧基活化,然后与nh2-ala-tyr(r5)-asp(r6)-oh进行液相偶联,得到r2-met-leu-val-ala-tyr(r5)-asp(r6)-oh。
在本发明的技术方案中,合成r1-ser(r4)-pro-met-leu-val-ala-tyr(r5)-asp(r6)-oh的方法为r2-met-leu-val-ala-tyr(r5)-asp(r6)-oh脱除氨基保护基r2,得到nh2-met-leu-val-ala-tyr(r5)-asp(r6)-oh;将r1-ser(r4)-pro-oh进行羧基活化,然后与nh2-met-leu-val-ala-tyr(r5)-asp(r6)-oh进行液相偶联,得到r1-ser(r4)-pro-met-leu-val-ala-tyr(r5)-asp(r6)-oh。
在本发明中,步骤4)脱除保护基的条件为以50%哌啶/dcm反应,再以tfa进行反应。
在本发明的技术方案中,r1、r2、r3、r7、r8分别独立地选自苄氧羰基(cbz)、叔丁氧羰基(boc)、笏甲氧羰基(fmoc)、烯丙氧羰基(alloc)、三甲基硅乙氧羰基(teoc);
r4选自tbu、trt,r5选自tbu,r6选自tbu。
在本发明的技术方案中,所述的羧基活化是将带有羧基的化合物原料以缩合剂进行羧基活化,其中的缩合剂选自dcc、hobt、hoat、edc、honb、hosu、dic、……。
在本发明的技术方案中,所述的液相偶联是在碱性条件下羧基活化的化合物与具有氨基基团的化合物在碱性条件下进行偶联。
在本发明的技术方案中,脱除氨基保护基的方法为以20%哌啶/dcm或50%哌啶/dcm进行处理或以tfa进行处理。
在本发明的技术方案中,纯化reltecimod的方法为以液相色谱方式进行纯化或者以纯结晶的方式进行纯化。
在本发明步骤5)中以hplc方式进行纯化,hplc的纯化条件为流动相a相为0.1%tfa,b相为乙腈,梯度:25%b—45%b。
在本发明一个具体的技术方案中,所述制备方法保护如下步骤:
步骤1):合成fmoc-ser(tbu)-pro-oh、fmoc-met-leu-val-oh和fmoc-ala-tyr(tbu)-asp(otbu)-oh三个全保护片段;
步骤2):fmoc-met-leu-val-oh和fmoc-ala-tyr(tbu)-asp(otbu)-oh连接,得到fmoc-met-leu-val-ala-tyr(tbu)-asp(otbu)-oh;
步骤3):将fmoc-ser(tbu)-pro-oh和fmoc-met-leu-val-ala-tyr(tbu)-asp(otbu)-oh连接,得到fmoc-ser(tbu)-pro-met-leu-val-ala-tyr(tbu)-asp(otbu)-oh;
步骤4:脱保护,得到reltecimod粗品;
步骤5:hplc制备,得到reltecimod纯品。
本发明采用液相方法,先合成1个二肽片段和2个3肽片段,之后按c端到n端的顺序依次将片段连接并脱保护即可得到粗品,粗品经hplc制备即可得到纯度99%以上,单杂小于0.1%纯品。
有益效果
本发明采用c端氨基酸刚性较大的片段进行合成,通过对片段质量进行控制和特定的连接顺序,使得目标产物中没有单个氨基酸缺省和内插的杂质,且几乎没有异构化杂质产生,得到粗品只需经过一步简单hplc制备即可得到高纯度的产品。
附图说明
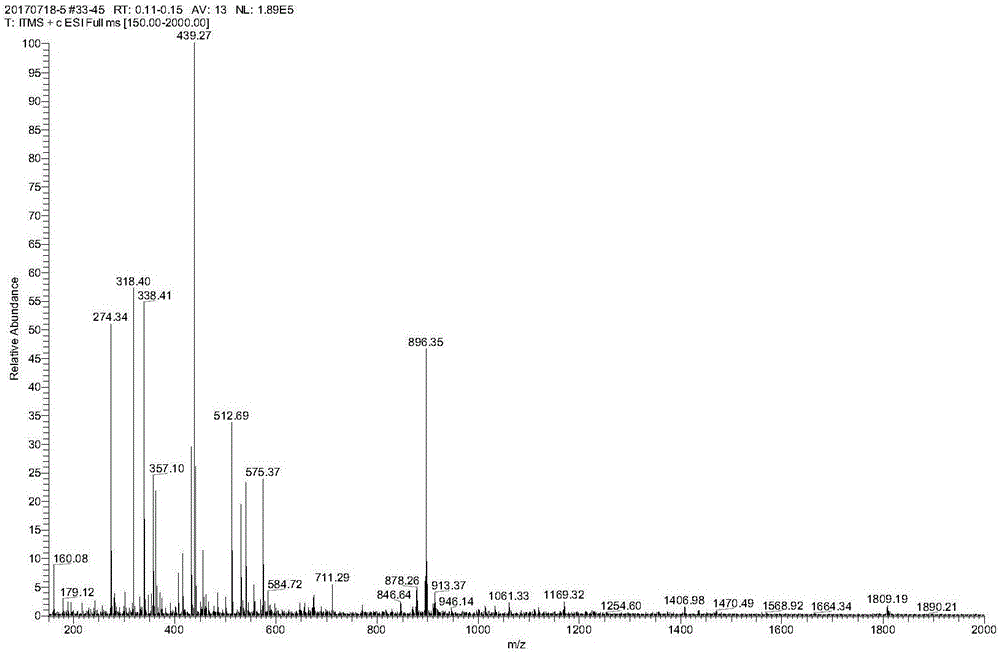
图1为实施例12所得纯品的质谱图。
图2为实施例12所得纯品的hplc谱图。
具体实施方式
下面结合具体的实施例来进一步阐述本发明。应理解,这些实施例仅用于说明本发明,而不是用来限制本发明的范围。下述实施例中所使用的实验方法如无特殊说明,均为常规方法。
实施例1:fmoc-ser(tbu)-pro-oh合成
将fmoc-ser(tbu)-oh(383.4g,1mol)、honb(197.1g,1.1mol)溶解于2升dcm中,0~5℃缓慢滴加dic(154.1g,1.2mol),滴加完毕后,反应液自然升温至室温,继续搅拌反应3h以上。反应液温度升至室温后,每隔1小时tlc(展开剂:pe:ea:acoh=2:1:0.03)检测一次反应,当tlc显示fmoc-ser(tbu)-oh反应完全时,停止搅拌。反应液过滤,减压浓缩后得到白色固体,固体加入2升ea搅拌洗涤0.5h,固体微溶。悬浊液0℃±2℃放置20~24h。产品减压过滤,et2o洗涤(544ml×2),真空干燥后得到470.1g白色固体fmoc-ser(tbu)-onb,收率:81.6%,纯度99.54%。
将h-pro-oh(172.7g,1.5mol)溶于4.5升乙腈-水(2:1)的混合溶剂中,加入上述fmoc-ser(tbu)-onb(470.1g,0.82mol),fmoc-ser(tbu)-onb分散于溶剂中,微溶。室温剧烈搅拌,缓慢滴加dipea(129.2g,1mol),滴加过程中fmoc-ser(tbu)-onb不断反应最后固体消失,之后室温搅拌2h以上。固体消失后每隔1小时tlc(展开剂:pe:ea:acoh=2:1:0.03)检测一次,当tlc显示fmoc-ser(tbu)-onb反应完全时,停止搅拌。旋干反应体系中的乙腈,残留溶液加入100ml10%碳酸钠水溶液,乙酸乙酯萃取(1l×3),水层用饱和柠檬酸酸化至ph3-4,再用乙酸乙酯萃取(1l×2),合并有机相,饱和食盐水洗涤(500ml×2),无水硫酸钠干燥。过滤,滤液真空浓缩,残留物中加入2let2o,加热至40℃溶解,静置至室温后2~8℃结晶20~24h。过滤,滤渣用冰冻无水乙醚洗涤2次,真空干燥后得到247g白色固体,收率:51.5%,纯度99.67%。
实施例2:boc-leu-val-oh合成
用1lthf溶解boc-leu-oh(231.3g,1mol)和honb(197.1g,1.1mol),冰浴条件下加入dcc(227.0g,1.1mol),室温搅拌4h,tlc显示boc-leu-oh反应完全,过滤除去固体,滤液备用。
用2l去离子水溶解na2co3(318g,3mol)、h-val-oh(117.8g,1mol),缓慢滴加上述新制备的滤液,室温搅拌18h,tlc显示boc-leu-onb反应完全,反应液真空浓缩掉thf,加入2l去离子水稀释残留液,ea提取(1l×2),水层用1mol/l盐酸溶液酸化至ph=3-4,ea萃取,合并ea层,无水硫酸钠快速干燥,过滤、真空浓缩,残留物用thf:ea:pe(1:2:10,v/v)重结晶。过滤收集晶体,真空干燥得到218.7gboc-leu-val-oh,收率:66.2%,纯度99.59%。
实施例3:fmoc-met-leu-val-oh合成
将boc-leu-val-oh(165.2g,0.5mol)溶解在1l三氟乙酸中,室温搅拌反应1h,tlc显示boc-leu-val-oh反应完全,真空浓缩掉tfa,得到的h-leu-val-oh油状物待用。
用0.5l乙腈溶解fmoc-met-oh(185.7g,0.5mol)和honb(98.6g,0.55mol),冰浴条件下加入dcc(227.0g,1.1mol),室温搅拌2h,tlc显示fmoc-met-oh反应完全,过滤除去dcu,得到的fmoc-met-onb滤液备用。
用0.5l去离子水溶解h-leu-val-oh油状物,饱和na2co3溶液调ph至8.0~8.5,缓慢滴加上述fmoc-met-onb溶液,滴加完毕后继续室温搅拌4h,tlc显示fmoc-met-onb反应完全。真空浓缩掉溶液中的thf,补加0.5l蒸馏水至剩余水溶液,ea萃取(0.5l×2),水相用饱和柠檬酸溶液调ph至3~4,ea萃取(0.5l×2),合并有机相,10%柠檬酸(0.5l×2)洗涤,饱和食盐水(0.5l×2)洗涤,无水na2so4干燥,真空浓缩,dcm/et2o结晶得到白色固体207.2g,收率71.0%,纯度99.75%。
实施例4:fmoc-tyr(tbu)-asp(otbu)-oh合成
将fmoc-tyr(tbu)-oh(459.5g,1mol)和honb(197.1g,1.1mol)溶解在thf(1l)中,冰浴条件下加入dcc(227.0g,1.1mol),搅拌反应2h,tlc显示原料反应完全,过滤,滤液待用。
将h-asp(otbu)-oh(208.1g,1.1mol)和na2co3(212g,2mol)溶解在1l蒸馏水中,室温搅拌下滴加上述滤液,滴加完毕后继续室温反应6h,tlc显示原料反应完全。真空浓缩掉溶液中的thf,补加1l蒸馏水至剩余水溶液,ea萃取(1l×2),水相用饱和柠檬酸溶液调ph至3~4,ea萃取(1l×2),合并有机相,10%柠檬酸(1l×2)洗涤,饱和食盐水(1l×2)洗涤,无水na2so4干燥,真空浓缩,ea/正己烷结晶得到白色固体425.1g,收率67.4%,纯度99.47%。
实施例5:h-tyr(tbu)-asp(otbu)-oh合成
将fmoc-tyr(tbu)-asp(otbu)-oh(315.4g,0.5mol)溶解在1l20%哌啶/dmf溶液中,室温搅拌0.5h,tlc显示原料反应。将反应液加入到5l10%柠檬酸冰水中,搅拌5min,过滤收集固体,水洗(1l×2),干燥后得到192.0g白色固体,收率:94.0%,纯度99.54%。
实施例6:fmoc-ala-tyr(tbu)-asp(otbu)-oh合成
将fmoc-ala-oh(124.5g,0.4mol)和honb(78.8g,0.44mol)溶解在thf(0.5l)中,冰浴条件下加入dcc(90.8g,0.44mol),搅拌反应2h,tlc显示原料反应完全,过滤,滤液待用。
将h-tyr(tbu)-asp(otbu)-oh(163.4g,0.4mol)和na2co3(106g,1mol)溶解在1l蒸馏水中,室温搅拌下滴加上述滤液,滴加完毕后继续室温反应4h,tlc显示原料反应完全。真空浓缩掉溶液中的thf,补加1l蒸馏水至剩余水溶液,ea萃取(1l×2),水相用饱和柠檬酸溶液调ph至3~4,ea萃取(1l×2),合并有机相,10%柠檬酸(1l×2)洗涤,饱和食盐水(1l×2)洗涤,无水na2so4干燥,真空浓缩,ea结晶得到白色固体227.9g,收率81.2%,纯度99.59%。
实施例7:h-ala-tyr(tbu)-asp(otbu)-oh合成
将fmoc-ala-tyr(tbu)-asp(otbu)-oh(140.4g,0.2mol)溶解在50%哌啶/dcm(300ml)中,室温搅拌0.5h,tlc显示原料反应完全。反应液缓慢加入2l冰冻无水乙醚中,过滤收集固体,冰冻无水乙醚洗涤(200ml×3),干燥,得到95.2g白色固体,收率95.1%,纯度99.48%。
实施例8:fmoc-met-leu-val-ala-tyr(tbu)-asp(otbu)-oh合成
将fmoc-met-leu-val-oh(116.7g,0.2mol)和honb(39.4g,0.22mol)溶解在thf(200ml)中,冰浴条件下加入dcc(45.4g,0.22mol),搅拌反应2h,tlc显示原料反应完全,过滤,滤液待用。
将h-ala-tyr(tbu)-asp(otbu)-oh(95.9g,0.2mol)和na2co3(42.4g,0.4mol)溶解在200ml蒸馏水中,室温搅拌下滴加上述滤液,滴加完毕后继续室温反应12h,tlc显示原料反应完全。真空浓缩掉溶液中的thf,补加200ml蒸馏水至剩余水溶液,ea萃取(200ml×3),水相用饱和柠檬酸溶液调ph至3~4,ea萃取(200l×2),合并有机相,水洗(200ml×3)洗涤,饱和食盐水(200ml×3)洗涤,无水na2so4干燥,真空浓缩,dmf/h2o结晶得到142.6g白色固体,收率68.2%,纯度99.42%。
实施例9:h-met-leu-val-ala-tyr(tbu)-asp(otbu)-oh合成
将fmoc-met-leu-val-ala-tyr(tbu)-asp(otbu)-oh(104.5g,100mmol)加入到500ml20%哌啶/dmf中,室温搅拌0.5h,tlc显示原料反应完全。反应液缓慢加入2.5l蒸馏水中,搅拌5min,过滤,水洗(500ml×3),干燥,得到白色固体78.3g,收率95.1%,纯度99.46%。
实施例10:fmoc-ser(tbu)-pro-met-leu-val-ala-tyr(tbu)-asp(otbu)-oh合成
将fmoc-ser(tbu)-pro-oh(24.0g,50mmol)和hosu(6.33g,55mmol)溶解在thf(50ml)中,冰浴条件下加入dcc(11.3g,55mmol),室温搅拌反应4h,tlc显示原料基本反应完全,过滤,滤液待用。
将h-met-leu-val-ala-tyr(tbu)-asp(otbu)-oh(41.2g,50mmol)和nahco3(16.8g,200mmol)溶解在100ml去离子水和50ml四氢呋喃的混合溶剂中,边搅拌边加入上述滤液,室温搅拌反应24h,tlc显示原料反应完全。真空浓缩掉溶剂,残留物中加入ea(50ml),搅拌0.5h,过滤,ea洗涤(50ml×2),10%柠檬酸洗(50ml×3),水洗(50ml×3)乙醚洗涤(50ml×3)干燥,得到35.0g白色固体,收率57.6%,纯度98.9%。
实施例11:reltecimod粗品合成
向装有fmoc-ser(tbu)-pro-met-leu-val-ala-tyr(tbu)-asp(otbu)-oh(12.2g,10mmol)的圆底烧瓶中加入到50%哌啶/dcm(100ml)溶液中,室温搅拌0.5h,hplc显示原料反应完全。将反应液加入到et2o(1l)中,搅拌0.5h,过滤,et2o洗涤(200ml×2),干燥,得到白色固体。
向装有上述固体的圆底烧瓶中加入tfa(95ml)和水(5ml)的混合溶液,室温搅拌3h,hplc显示原料反应完全。将反应液加入et2o(1l)中,离心收集固体,et2o洗涤(200ml×3),真空干燥得到8.63g白色固体,收率98.0%,纯度95.7%。
实施例12:hplc制备reltecimod纯品
样品处理:取2greltecimod粗品,加入25%乙腈/水溶液(200ml),超声使样品完全溶解后用滤膜过滤,收集滤液备用。
hplc纯化条件:色谱柱固定相为十八烷基硅烷键合硅胶,柱子直径和长度为50mm×250mm。流动相a相为0.1%tfa,b相为乙腈,流速为80ml/min,梯度:25%b—45%b,检测波长,280nm。
纯化过程:色谱柱平衡5min后上样,运行梯度纯化,监测并收集目的峰馏分。将目的峰馏分减压浓缩至30ml后冻干。冻干后得白色粉末状固体纯品1.68g。纯度99.82%,单个杂质均小于0.1%。收率84.0%,ms:896.35(m+1)。
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此理解为对本发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
- 还没有人留言评论。精彩留言会获得点赞!