一种对引物测序的方法与流程
本发明涉及基因检测鉴定技术领域,特别涉及一种对引物测序的方法。
背景技术:
引物(primer),是一小段单链dna或rna,作为dna复制的起始点,在核酸合成反应时,作为每个多核苷酸链进行延伸的出发点而起作用的多核苷酸链,在引物的3′-oh上,核苷酸以二酯链形式进行合成,因此引物的3′-oh,必须是游离的。之所以需要引物是因为在dna合成中dna聚合酶仅仅可以把新的核苷酸加到已有的dna链上。
目前引物合成主要采用固相亚磷酰胺y三酯法,该方法具有高效、快速的偶联以及起始反应物比较稳定的特点。该方法主要是将dna固定在固相载体上完成dna链的合成,由待合成引物的3'端向5'端合成延伸,相邻的核苷酸通过3'→5'磷酸二酯键连接。
现有技术中,固相亚磷酰胺三酯法合成引物的具体步骤如下:
1)用三氯乙酸去除固相载体5'-羟基的保护基团dmt,获得游离的5'-羟基;
2)将亚磷酰胺保护核苷酸单体与活化剂四氮唑混合,得到核苷亚磷酸活化中间体,与游离的5'-羟基发生缩合反应;
3)由于无法保证百分之百的5'-羟基都参与缩合,可能有极少数5'-羟基没有参加反应(少于2%),可用乙酸酐和1-甲基咪唑试剂将其终止合成,这种短片段可以在纯化时分离掉;
4)在氧化剂的作用下,亚磷酰形式转变为更稳定的磷酸三酯,使dna磷酸骨架更稳定。
经过以上四个步骤,一个脱氧核苷酸就被连接到固相载体上;重复以上步骤,直到所有要求合成的碱基连接完成;合成过程中可以监控脱下的保护基团dmt的颜色可以初步判定合成效率。
在此基础上,为了确定人工合成的引物序列的碱基排列是否正确,现有技术中的引物检测方法主要包括:高效液相色谱法(hplc),毛细管凝胶电泳法(cge),以及质谱分析(ms)。其中,hplc是一种高效的分析寡核苷酸的方法;cge是一种将凝胶移到毛细管中作支持物进行的电泳,基于尺寸排阻机理,可实现单核苷酸分离,可用于测定寡核苷酸纯度,分辨率高,定量准确;ms是用电场和磁场将运动的离子(带电荷的原子、分子或分子碎片)按它们的质荷比分离后进行检测的方法,ms能够精确地测定引物的分子量大小。
然而,事实上,上述各方法中都存在不容忽视的技术缺陷,例如,虽然质谱能够精确检测序列的总分子量,却也仅能知道所检测引物的总分子量,而无法识别其碱基序列排列顺序,更不能识别个别碱基的突变;又如,hplc和cge可检测引物主片段所占比例,但是主要是针对引物的纯度进行检测,无法识别序列的长短及具体的碱基排列。因此,现有技术中尚未提供一种能够精确鉴定合成引物的碱基序列的方法。
技术实现要素:
为了克服现有技术中存在的种种技术缺陷,本发明旨在提供一种全新的对引物本身进行测序的方法,该方法既能识别序列的长短与总分子量,又能精确鉴定碱基序列(核苷酸序列)的排列顺序。
具体地,本发明提供了一种对引物测序的方法,其包括以下步骤:
步骤一:在扩增体系中,以引物z和引物y2作为pcr引物对,将质粒模板dna经pcr扩增出模板dna1,然后采用琼脂糖凝胶电泳法回收模板dna1;
步骤二:在扩增体系中,以引物x和引物y2作为pcr引物对,将模板dna1经pcr扩增出待测dna2,然后采用琼脂糖凝胶电泳法回收待测dna2;
步骤三:将待测dna2的原液稀释至浓度为2.5~5ng/ul,然后,使用引物y2对待测dna2进行sanger法测序,即测得引物x的序列;
其中,所述引物x为待测序的引物;所述引物y2为一条已知的通用引物;所述引物z由所述引物x的3'端连接引物y1构成,所述引物y1为另一条已知的通用引物。
值得说明的是,所述通用引物例如可为:1492r,27f,3.0rev,35s,3'ad,3'aox,3'bd,m13-47,m13r,rv-m,male中的任一序列。因此,上述引物y1与引物y2在序列的选择方面不存在特定的要求或相互联系,只要是已知的通用引物,尤其是基于质粒的通用引物,均可被采用。
优选地,在上述对引物测序的方法中,所述模板dna1为200~700bp的dna片段。
优选地,在上述对引物测序的方法中,所述待测dna2为700~800bp的dna片段。
优选地,在上述对引物测序的方法中,所述步骤一中的扩增体系包含:
优选地,在上述对引物测序的方法中,所述步骤二中的扩增体系包含:
其中,2×easytaqpcrsupermix(+dye)为现有的市售产品,其包含
此外,在所述步骤二中,“模板dna1(原液稀释100倍)”是指将模板dna1的原液稀释100倍后,取1ul,与引物x、引物y2等组成该扩增体系。并且,本文记载的括号中的“10um”,“150ng/ul”等均表示相关物质的浓度;例如,引物y2(10um)则表示浓度为10um的引物y2。
优选地,在上述对引物测序的方法中,所述步骤一和所述步骤二中各自实施的pcr扩增的反应程序为:
其中,“循环30次”是指依次将变性、退火与延伸过程循环30次;由于这些表示方式和相应的操作步骤都是本技术领域的研究人员所熟知的,因此在本文中不再赘述。
优选地,在上述对引物测序的方法中,在所述步骤一采用的琼脂糖凝胶电泳法中,琼脂糖凝胶的质量体积浓度为0.8%。
优选地,在上述对引物测序的方法中,在所述步骤二采用的琼脂糖凝胶电泳法中,琼脂糖凝胶的质量体积浓度为1.8%。
其中,琼脂糖凝胶电泳法的具体操作步骤是本领域的已知方法,因此,在本文中不再赘述。
综上所述,发明人将引物z设计为x+y1,先利用引物z和引物y2制备模板dna1,接着利用引物x和引物y2制备待测dna2,最后使用引物y2对待测dna2进行sanger法测序,成功测得引物x的完整序列,从而达到了检测合成的引物序列的碱基排列是否正确的目的。因此,与现有的引物测序方法相比,本发明所提供的对引物测序的方法具有以下有益的技术效果:既能识别待测序的引物的序列长短与总分子量,又能精确地鉴定待测序的引物的序列中的每一个碱基,其检测结果百分百准确,这是因为该引物的核苷酸序列(碱基序列)会被逐个碱基地完成测定;并且,所述对引物测序的方法的各步骤操作简单;因此,所述对引物测序的方法具有广阔的应用前景,例如可用于药物研发、生物诊断试剂研发、基因工程等相关技术领域。
附图说明
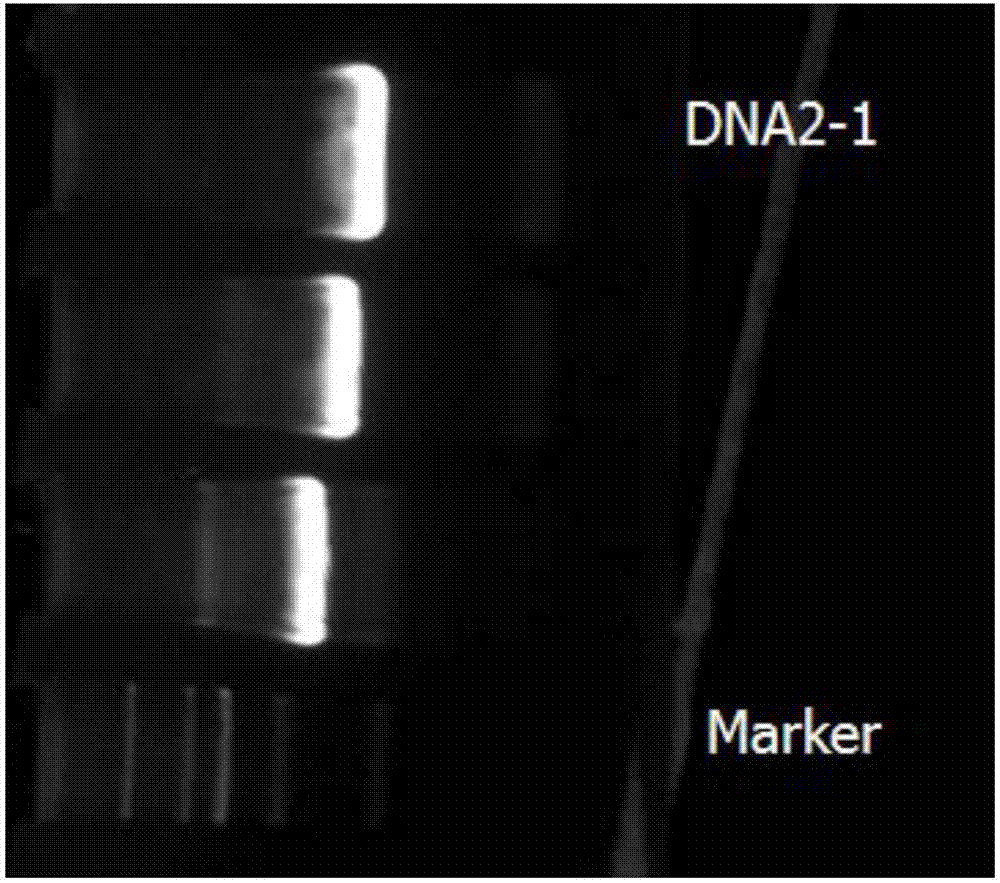
图1为实施例1的步骤一中测得的各dna的琼脂糖凝胶电泳图;
图2为实施例1的步骤二中测得的各dna的琼脂糖凝胶电泳图;
图3为实施例1的步骤三中的dna2-1的sanger法测序图,其中显示了引物x-1的反向互补序列;
图4为实施例2的步骤一中测得的各dna的琼脂糖凝胶电泳图;
图5为实施例2的步骤二中测得的各dna的琼脂糖凝胶电泳图;
图6为实施例2的步骤三中的dna2-2的sanger法测序图,其中显示了引物x-2的反向互补序列;
图7为实施例3的步骤一中测得的各dna的琼脂糖凝胶电泳图;
图8为实施例3的步骤二中测得的各dna的琼脂糖凝胶电泳图;
图9为实施例3的步骤三中的dna2-3的sanger法测序图,其中显示了引物x-3的反向互补序列;
图10为实施例4的步骤一中测得的各dna的琼脂糖凝胶电泳图;
图11为实施例4的步骤二中测得的各dna的琼脂糖凝胶电泳图;
图12为实施例4的步骤三中的dna2-4的sanger法测序图,其中显示了引物x-4的反向互补序列。
具体实施方式
下面结合具体实施方式对本发明作进一步阐述,但本发明并不限于以下实施方式。
根据本发明的第一方面的一种对引物测序的方法,包括以下步骤:
步骤一:在扩增体系中,以引物z和引物y2作为pcr引物对,将质粒模板dna经pcr扩增出模板dna1,然后采用琼脂糖凝胶电泳法回收模板dna1;
步骤二:在扩增体系中,以引物x和引物y2作为pcr引物对,将模板dna1经pcr扩增出待测dna2,然后采用琼脂糖凝胶电泳法回收待测dna2;
步骤三:将待测dna2的原液稀释至浓度为2.5~5ng/ul,然后,使用引物y2对待测dna2进行sanger法测序,即测得引物x的序列;
其中,所述引物x为待测序的引物;所述引物y2为一条已知的通用引物;所述引物z由所述引物x的3'端连接引物y1构成,所述引物y1为另一条已知的通用引物。
在一个优选实施例中,所述模板dna1为200~700bp的dna片段。
在一个优选实施例中,所述待测dna2为700~800bp的dna片段。
在一个优选实施例中,所述步骤一中的扩增体系包含:
在一个优选实施例中,所述步骤二中的扩增体系包含:
在一个优选实施例中,所述步骤一和所述步骤二中各自实施的pcr扩增的反应程序为:
在一个优选实施例中,在所述步骤一采用的琼脂糖凝胶电泳法中,琼脂糖凝胶的质量体积浓度为0.8%。
在一个优选实施例中,在所述步骤二采用的琼脂糖凝胶电泳法中,琼脂糖凝胶的质量体积浓度为1.8%。
以下实施例1~4中所使用的引物的核苷酸序列如下表1所示:
表1
实施例1
使用plasmid1(通用质粒)为模板,最终扩增得到的dna2-1片段长度约为353bp;
待测序的引物x-1的核苷酸序列为:
seqidno:7;
已知的通用引物y1-1的核苷酸序列为:
seqidno:1;
已知的通用引物y2-1的核苷酸序列为:
seqidno:2;
引物z-1的核苷酸序列为:
seqidno:11。
步骤一(制备dna1-1):
将plasmid1模板dna经pcr扩增出dna1-1,然后采用琼脂糖凝胶电泳法回收dna1-1;其中,扩增体系包含:
其中,pcr扩增的反应程序为:
并且,琼脂糖凝胶电泳法中的琼脂糖凝胶的质量体积浓度为0.8%,上样量为5ul;分离检测结果如图1所示,其中,2kbmarker条带大小依次为:2000,1000,750,500,250,100bp。
步骤二(制备dna2-1):
将dna1-1经pcr扩增出待测dna2-1,然后采用琼脂糖凝胶电泳法回收待测dna2-1;其中,扩增体系包含:
其中,pcr扩增的反应程序为:
并且,琼脂糖凝胶电泳法中的琼脂糖凝胶的质量体积浓度为1.8%,上样量为30ul,切胶回收;分离检测结果如图2所示,其中,2kbmarker条带大小依次为:2000,1000,750,500,250,100bp。
步骤三(sanger法测序):
将待测dna2-1的原液稀释至浓度为2.5ng/ul,然后,使用引物y2-1对待测dna2-1进行sanger法测序,测序结果如图3所示,显示了以下序列(包含引物x-1的反向互补序列):seqidno:15。
从而完成了对引物x-1的测序。
实施例2
使用plasmid2(通用质粒)为模板,最终扩增得到的dna2-2片段长度约为380bp;
待测序的引物x-2的核苷酸序列为:
seqidno:8;
已知的通用引物y1-2的核苷酸序列为:
seqidno:3;
已知的通用引物y2-2的核苷酸序列为:
seqidno:4;
引物z-2的核苷酸序列为:
seqidno:12。
步骤一(制备dna1-2):
将plasmid2模板dna经pcr扩增出dna1-2,然后采用琼脂糖凝胶电泳法回收dna1-2;其中,扩增体系包含:
其中,pcr扩增的反应程序为:
并且,琼脂糖凝胶电泳法中的琼脂糖凝胶的质量体积浓度为0.8%,上样量为5ul;分离检测结果如图4所示,其中,2kbmarker条带大小依次为:2000,1000,750,500,250,100bp。
步骤二(制备dna2-2):
将dna1-2经pcr扩增出待测dna2-2,然后采用琼脂糖凝胶电泳法回收待测dna2-2;其中,扩增体系包含:
其中,pcr扩增的反应程序为:
并且,琼脂糖凝胶电泳法中的琼脂糖凝胶的质量体积浓度为1.8%,上样量为30ul,切胶回收;分离检测结果如图5所示,其中,2kbmarker条带大小依次为:2000,1000,750,500,250,100bp。
步骤三(sanger法测序):
将待测dna2-2的原液稀释至浓度为2.5ng/ul,然后,使用引物y2-2对待测dna2-2进行sanger法测序,测序结果如图6所示,显示了以下序列(包含引物x-2的反向互补序列):seqidno:16。
从而完成了对引物x-2的测序。
实施例3
使用plasmid3(通用质粒)为模板,最终扩增得到的dna2-3片段长度约为453bp;
待测序的引物x-3的核苷酸序列为:
seqidno:9;
已知的通用引物y1-3的核苷酸序列为:
seqidno:5;
已知的通用引物y2-3的核苷酸序列为:
seqidno:6;
引物z-3的核苷酸序列为:
seqidno:13。
步骤一(制备dna1-3):
将plasmid3模板dna经pcr扩增出dna1-3,然后采用琼脂糖凝胶电泳法回收dna1-3;其中,扩增体系包含:
其中,pcr扩增的反应程序为:
并且,琼脂糖凝胶电泳法中的琼脂糖凝胶的质量体积浓度为0.8%,上样量为5ul;分离检测结果如图7所示,其中,2kbmarker条带大小依次为:2000,1000,750,500,250,100bp。
步骤二(制备dna2-3):
将dna1-3经pcr扩增出待测dna2-3,然后采用琼脂糖凝胶电泳法回收待测dna2-3;其中,扩增体系包含:
其中,pcr扩增的反应程序为:
并且,琼脂糖凝胶电泳法中的琼脂糖凝胶的质量体积浓度为1.8%,上样量为30ul,切胶回收;分离检测结果如图8所示,其中,2kbmarker条带大小依次为:2000,1000,750,500,250,100bp。
步骤三(sanger法测序):
将待测dna2-3的原液稀释至浓度为2.5ng/ul,然后,使用引物y2-3对待测dna2-3进行sanger法测序,测序结果如图9所示,显示了以下序列(包含引物x-3的反向互补序列):seqidno:17。
从而完成了对引物x-3的测序。
实施例4
使用plasmid1(通用质粒)为模板,最终扩增得到的dna2-4片段长度约为790bp;
待测序的引物x-4的核苷酸序列为:
seqidno:10;
已知的通用引物y1-1的核苷酸序列为:
seqidno:1;
已知的通用引物y2-1的核苷酸序列为:
seqidno:2;
引物z-4的核苷酸序列为:
seqidno:14。
步骤一(制备dna1-4):
将plasmid1模板dna经pcr扩增出dna1-4,然后采用琼脂糖凝胶电泳法回收dna1-4;其中,扩增体系包含:
其中,pcr扩增的反应程序为:
并且,琼脂糖凝胶电泳法中的琼脂糖凝胶的质量体积浓度为0.8%,上样量为5ul;分离检测结果如图10所示,其中,2kbmarker条带大小依次为:2000,1000,750,500,250,100bp。
步骤二(制备dna2-4):
将dna1-4经pcr扩增出待测dna2-4,然后采用琼脂糖凝胶电泳法回收待测dna2-4;其中,扩增体系包含:
其中,pcr扩增的反应程序为:
并且,琼脂糖凝胶电泳法中的琼脂糖凝胶的质量体积浓度为1.8%,上样量为30ul,切胶回收;分离检测结果如图11所示,其中,2kbmarker条带大小依次为:2000,1000,750,500,250,100bp。
步骤三(sanger法测序):
将待测dna2-4的原液稀释至浓度为5ng/ul,然后,使用引物y2-1对待测dna2-4进行sanger法测序,测序结果如图12所示,显示了以下序列(包含引物x-4的反向互补序列):seqidno:18。
从而完成了对引物x-4的测序。
此外,以上实施例1~4中sanger法测序的dna片段的核苷酸序列如下表2所示:
表2
以上对本发明的具体实施例进行了详细描述,但其只是作为范例,本发明并不限制于以上描述的具体实施例。对于本领域技术人员而言,任何对本发明进行的等同修改和替代也都在本发明的范畴之中。因此,在不脱离本发明的精神和范围下所作的均等变换和修改,都应涵盖在本发明的范围内。
序列表
<110>北京睿博兴科生物技术有限公司
<120>一种对引物测序的方法
<160>18
<170>siposequencelisting1.0
<210>1
<211>18
<212>dna
<213>人工序列(artificialsequence)
<400>1
tgtaaaacgacggccagt18
<210>2
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>2
agcggataacaatttcacacagga24
<210>3
<211>25
<212>dna
<213>人工序列(artificialsequence)
<400>3
tgtaaaacgacggccagtgtctgag25
<210>4
<211>18
<212>dna
<213>人工序列(artificialsequence)
<400>4
caggaaacagctatgacc18
<210>5
<211>31
<212>dna
<213>人工序列(artificialsequence)
<400>5
tgtaaaacgacggccagtgtctgaggctcgc31
<210>6
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>6
gtcaatactgacgatggtca20
<210>7
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>7
agctacatcagcacattcat20
<210>8
<211>30
<212>dna
<213>人工序列(artificialsequence)
<400>8
ggatcctcaatcactgtcgtgctcagcatc30
<210>9
<211>40
<212>dna
<213>人工序列(artificialsequence)
<400>9
caccaagctcgacgaggtgaaggcctctcagcttaccttc40
<210>10
<211>46
<212>dna
<213>人工序列(artificialsequence)
<400>10
ggatcttcggctgcgatcaacgttttagagctagaaatagcaagtt46
<210>11
<211>38
<212>dna
<213>人工序列(artificialsequence)
<400>11
agctacatcagcacattcattgtaaaacgacggccagt38
<210>12
<211>55
<212>dna
<213>人工序列(artificialsequence)
<400>12
ggatcctcaatcactgtcgtgctcagcatctgtaaaacgacggccagtgtctgag55
<210>13
<211>71
<212>dna
<213>人工序列(artificialsequence)
<400>13
caccaagctcgacgaggtgaaggcctctcagcttaccttctgtaaaacgacggccagtgt60
ctgaggctcgc71
<210>14
<211>64
<212>dna
<213>人工序列(artificialsequence)
<400>14
ggatcttcggctgcgatcaacgttttagagctagaaatagcaagtttgtaaaacgacggc60
cagt64
<210>15
<211>315
<212>dna
<213>人工序列(artificialsequence)
<400>15
gatcatgtgcccaaaccctctgcaacgatggcactgaatatcccgtgtacgtcctgtgaa60
gaagcggcgcctttggcagggggcgccaccggcttggtcgtccctgtgcgcgatgtagtg120
ttaggcgtagaaggtgcagggctggaataacatcgcaatcagcataatcagaataagaac180
ccagcgaaaaggggacacgtaccgaacgtgttacctttattttaccaccacaaagggcag240
cttcaattcgccctatagtgagtcgtattacaattcactggccgtcgttttacaatgaat300
gtgctgatgtagcta315
<210>16
<211>349
<212>dna
<213>人工序列(artificialsequence)
<400>16
cgacttcgcgtgtcgccctttctgcttacgtgccttcaccacctgtatggcacggctgtg60
gtacgcgtcgttgttttccttgttgtactcgctcatggcgaagttcagcgcttccaacac120
tccggtgtcgttggcgtttgcatcaatcacgccgcccaccaagcgcataccttgcttggg180
ggacgcggcccaggccaccgccaggacagccagcaggagcagcagggatcgtagtgggct240
agccataagggcgacacgcgaatacgataacaagcatcaggactgaagcgagcctcagac300
actggccgtcgttttacagatgctgagcacgacagtgattgaggatcca349
<210>17
<211>427
<212>dna
<213>人工序列(artificialsequence)
<400>17
cgatatcgaattcgcgtgtcgccctttcctctgtatgattcgcttgctccaaacgaaaca60
tatgtgaggagttgtcctccggcatgatgcgattgagaggcagaatccaggagctttggt120
gcggctgccaggcggcgtccgggctcttggcagggatgccgagcatctccatgccggact180
ggagcccgacgagctcgtgggtctgaggcgagaactcgccatacccgaatccttcatgat240
ccattgagacgatctgacaaacggttttgggagagcagctagcaagggattgaggactcg300
ctacaaagcttcataagggcgacacgcgaattcgatatcaagcttcaggactgcagcgag360
cctcagacactggccgtcgttttacagaaggtaagctgagaggccttcacctcgtcgagc420
ttggtga427
<210>18
<211>751
<212>dna
<213>人工序列(artificialsequence)
<400>18
gctcggatcactagtaacggccgccagtgtgctggaattgcccttagcctttcagcagag60
gtccaatcaaatggcctctagcccagcatgcagaacagaatttctcttacctcttagacg120
gcatcctggatgagctgctgcacgtccacctcctgcttcctcagggttccaaacgaagag180
tgactctccaccacgcctttgccaccaacgttcagcttgaccttccccacgactgtccca240
atagccccgctcttatggttctcacacttgctctcgtatttcacgaagtctgactccaca300
accactggcaagaaaatagtagatgaatctgaaggcagaggggctacctcagttgtcatg360
ggcagcacagcttcccactagcctcttgggaaacccataccagcatccaattggaatgtg420
gacatctggggttagtaaaatgcagtctacacaactatatgctgacagagtatagtcctg480
gactggccagagcctaggggtgattagcccctctttagccacagtaatggttaatattga540
ctgccaacctgaaaggatctagaatcgcctaggagacagacctcaggataaagggcaatt600
ctgcagatatccatcacactggcggccgctcgagcatgcatctagagggcccaattcgcc660
ctatagtgagtcgtattacaattcactggccgtcgttttacaaacttgctatttctagct720
ctaaaacgttgatcgcagccgaagatccaaa751
- 还没有人留言评论。精彩留言会获得点赞!