用于鉴定嵌合产物的条形码化环状文库构建的制作方法
本发明涉及核酸测序的领域。更具体地,本发明涉及产生用于核酸测序的条形码化模板文库的领域。
发明背景
当前一代的核酸测序方法利用靶分子的文库,由此对每个单独分子进行测序。文库中的每个分子包含待分析的靶序列,其与选择的测序方法和测序仪器所必需的人工序列缀合。人工序列通常包括条形码,用于独特地标记单独分子或分子组的短核苷酸序列。
独特分子条形码具有多种用途。标记和追踪每个单独的核酸分子使得能够检测极端罕见的序列,例如,在患者血液中以痕量存在和用于癌症的非侵入性早期检测和精确监测的循环肿瘤dna(ctdna)(参见newman,a.,等人,(2014)anultrasensitivemethodforquantitatingcirculatingtumordnawithbroadpatientcoverage,naturemedicinedoi:10.1038/nm.3519.)。单一靶分子的整个后代用相同的条形码标记并形成条形码化家族。因此,条形码可用于错误校正。没有被条形码化家族的所有成员共享的序列变化作为伪像而非真正的突变被丢弃。条形码也可用于位置去重复(positionaldeduplication)和靶标定量,因为整个家族代表原始样品中的单一分子(参见newman,a.,等人,(2016)integrateddigitalerrorsuppressionforimproveddetectionofcirculatingtumordna,naturebiotechnology34:547)。
条形码实现的错误校正已大大增强测序测定的灵敏度。测序伪像诸如聚合酶错误不再是检测罕见点突变的障碍。同时,条形码对于检测易位(基因融合)(人恶性肿瘤中的另一种常见类型的突变)没有那么有益。参见f.mertens,等人(2015)theemergingcomplexityofgenefusionsincancer,nat.rev.cancer15:371;f.mitelman,等人(2007)theimpactoftranslocationsandgenefusionsoncancercausation,nat.rev.cancer7:233。由于条形码通常被随机连接至靶分子的两个末端,所以不知道哪些5'条形码最初与哪些3'条形码相关。这在文库制备的扩增步骤期间产生问题,因为嵌合分子经由pcr中的模板转换产生。存在于测序文库中的人工产生的嵌合分子与可能已存在于原始样品中的真实基因融合体不可区分。这直接限制检测低频基因融合体(这可以是癌症中重要的驱动突变)的能力。需要一种基于条形码的方法来追踪和消除人工基因融合体,以使得能够检测真正的突变。
发明概述
在一些实施方案中,本发明是从包含多种靶分子的样品制备靶核酸分子的文库的方法,所述方法对于基本上每种靶分子包括:将单一衔接子连接至靶分子,形成环状分子,其中所述衔接子包含两个条形码,位于两个条形码之间的两个引物结合位点,其中退火至所述结合位点的引物彼此背离,和位于两个引物结合位点之间的至少一个修饰的核苷酸,其实现核酸聚合酶的链合成的终止;使与所述衔接子互补的正向引物退火至所述靶分子的一条链;将所述正向引物延伸直至所述修饰的核苷酸,由此产生第一链;使与所述衔接子互补的反向引物退火至所述第一链;延伸所述第一引物,由此产生第二链和包含第一链和第二链的双链分子,其中两个条形码侧接所述靶序列。在一些实施方案中,正向引物和反向引物中的至少一个包含与所述衔接子不互补且包含额外引物结合位点的5'-襟翼序列(flapsequence)。然后,所述方法进一步包括以下步骤:使额外引物退火至与正向引物中的襟翼序列互补的序列且延伸所述额外引物,由此产生包含两个额外引物位点和侧接所述靶序列的两个条形码的双链分子。在一些实施方案中,所述靶分子和所述衔接子是单链的。在其他实施方案中,所述靶分子和所述衔接子是双链的,且所述环状分子在引物的退火前至少部分变性。在一些实施方案中,所述条形码是4-20个碱基长的核苷酸序列。实现核酸聚合酶的链合成的终止的修饰的核苷酸可以选自无碱基核苷酸、具有蛋白侧基的核苷酸、合成核苷酸arac(阿糖胞苷)或脱氧尿嘧啶、异鸟嘌呤、5-甲基异胞嘧啶、乙二醇间隔基、具有大体积类似物诸如荧光团的核苷酸或非天然碱基对(ubp)“d5sics-dnam”核酸类似物。所述连接可以选自突出端连接、t-a连接、平末端连接和拓扑异构酶催化的连接。在一些实施方案中,所述衔接子在一个末端具有光可裂解接头。在这些实施方案中,所述接头在一个末端连接并暴露于uv光以使得能够在另一个末端连接。在一些实施方案中,所述额外引物是测序引物。
在一些实施方案中,本发明是靶核酸分子的文库,其中每个分子是环状分子,其包含靶序列和连接靶序列的末端的衔接子,所述衔接子包含:两个条形码;位于两个条形码之间的两个引物结合位点,其中退火至所述结合位点的引物彼此背离;位于两个引物结合位点之间的至少一个修饰的核苷酸,其实现核酸聚合酶的链合成的终止。在一些实施方案中,所述条形码是4-20个碱基长的核苷酸序列。实现核酸聚合酶的链合成的终止的修饰的核苷酸可以选自无碱基核苷酸、具有蛋白侧基的核苷酸、合成核苷酸arac(阿糖胞苷)或脱氧尿嘧啶、异鸟嘌呤、5-甲基异胞嘧啶、乙二醇间隔基、具有大体积类似物诸如荧光团的核苷酸或非天然碱基对(ubp)“d5sics-dnam”核酸类似物。
在其他实施方案中,本发明是对包含多种靶分子的样品中的靶核酸进行测序的方法,所述方法包括:通过将单一双链衔接子连接至基本上每种双链靶分子、形成双链环状分子,从所述样品产生靶核酸分子的文库,其中所述衔接子包含两个条形码,位于两个条形码之间的两个引物结合位点,其中退火至所述结合位点的引物彼此背离,和位于两个引物结合位点之间的至少一个修饰的核苷酸,其实现核酸聚合酶的链合成的终止;使所述双链环状靶分子的至少一部分变性;使与所述衔接子互补的正向引物退火至所述靶分子的一条链;将所述正向引物延伸直至所述修饰的核苷酸,由此产生第一链;使与所述衔接子互补的反向引物退火至所述第一链;延伸所述第一引物,由此产生第二链和包含第一链和第二链的双链分子,其中两个条形码侧接所述靶序列;扩增所述双链分子;和对所述双链分子的扩增产物进行测序。在一些实施方案中,正向引物和反向引物中的至少一个包含与所述衔接子不互补且包含额外引物结合位点的5'-襟翼序列。在一些实施方案中,所述方法在延伸第一引物之后进一步包括,使额外引物退火至与正向引物中的襟翼序列互补的序列且延伸所述额外引物,由此产生包含两个额外引物位点和侧接所述靶序列的两个条形码的双链分子。在一些实施方案中,可以用额外引物进行扩增或测序。
附图简述
图1是根据本发明的单链条形码化文库分子的图。
图2是用正向引物的第一链合成起始的图。
图3是第一链合成和终止的图。
图4是完成的第一链的图。
图5是使用第一链作为模板,用反向引物的第二链合成起始的图。
图6是完成的第二链的图。
图7是使用第二链作为模板,用正向引物的下一轮第一链合成起始的图。
图8是完成的测序模板分子的图。
发明详述
定义
以下定义有助于理解本公开。
术语“样品”是指含有或假定含有靶核酸的任何组合物。这包括从个体分离的组织或液体样品,例如皮肤、血浆、血清、脊髓液、淋巴液、滑液、尿液、泪液、血细胞、器官和肿瘤,以及由取自个体患者或模型生物体的细胞建立的体外培养物的样品,包括福尔马林固定的石蜡包埋的组织(ffpet)和从其分离的核酸。样品还可以包括无细胞材料,诸如含有无细胞dna(cfdna)或循环肿瘤dna(ctdna)的无细胞血液级分。
术语“核酸”是指核苷酸(例如,天然和非天然的核糖核苷酸和脱氧核糖核苷酸)的聚合物,包括dna、rna及其亚类,诸如cdna、mrna等。核酸可以是单链或双链的并且将通常含有5'-3'磷酸二酯键,尽管在一些情况下,核苷酸类似物可以具有其他键。核酸可以包括天然存在的碱基(腺苷、鸟苷、胞嘧啶、尿嘧啶和胸苷)以及非天然碱基。非天然碱基的一些实例包括在例如seela等人,(1999)helv.chim.acta82:1640中描述的那些。非天然碱基可以具有特定功能,例如,增加核酸双链体的稳定性,抑制核酸酶消化或阻断引物延伸或链聚合。
术语“多核苷酸”和“寡核苷酸”可互换使用。多核苷酸是单链或双链核酸。寡核苷酸是有时用于描述较短多核苷酸的术语。寡核苷酸可以由至少6个核苷酸或约15-30个核苷酸构成。寡核苷酸通过本领域已知的任何合适的方法,例如,通过如以下中所述的涉及直接化学合成的方法来制备:narang等人(1979)meth.enzymol.68:90-99;brown等人(1979)meth.enzymol.68:109-151;beaucage等人(1981)tetrahedronlett.22:1859-1862;matteucci等人(1981)j.am.chem.soc.103:3185-3191。
术语“引物”是指与靶核酸中的序列(“引物结合位点”)杂交且能够在适合于这种合成的条件下充当沿着核酸的互补链的合成的起始点的单链寡核苷酸。所述引物结合位点对于每种靶标可以是独特的,或者可以添加至所有靶标(“通用引发位点”或“通用引物结合位点”)。
术语“衔接子”意指可以被添加至另一序列、以便将额外的特性输入该序列的核苷酸序列。衔接子通常是这样的寡核苷酸,其可以是单链或双链的,或者可以具有单链部分和双链部分两者。衔接子可以含有序列,诸如条形码和通用引物或探针位点。
术语“连接”是指接合两条核酸链的缩合反应,其中一个分子的5'-磷酸基团与另一个分子的3'-羟基反应。连接通常是由连接酶或拓扑异构酶催化的酶促反应。连接可以接合两条单链以产生一个单链分子。连接还可以接合各自属于一个双链分子的两条链,因此接合两个双链分子。连接还可以将双链分子的两条链接合至另一双链分子的两条链,因此接合两个双链分子。连接还可以接合双链分子内的链的两个末端,因此修复双链分子中的切口。
术语“条形码”是指可以检测和标识的核酸序列。条形码可以被并入各种核酸中。条形码足够长,例如2、5、10个核苷酸,使得在样品中,并入条形码的核酸可以根据条形码区分或分组。
术语“多重标识符”和“mid”是指标识靶核酸来源(例如,衍生出所述核酸的样品,当组合来自多种样品的核酸时,其是所需要的)的条形码。来自相同样品的所有或基本上所有靶核酸将共享相同的mid。可以将来自不同来源或样品的靶核酸混合并同时测序。使用mid,可以将序列读数分配至靶核酸起源的单独样品。
术语“独特分子标识符”和“uid”是指标识与其连接的核酸的条形码。来自相同样品的所有或基本上所有靶核酸将具有不同的uid。源自相同原始靶核酸的所有或基本上所有后代(例如,扩增子)将共享相同的uid。
术语“通用引物”和“通用引发结合位点”或“通用引发位点”是指存在于(通常,体外添加至)不同靶核酸的引物和引物结合位点。例如,所述通用引发位点可以包括在与多个靶核酸连接的衔接子中。所述通用引发位点也可以是靶标特异性(非通用)引物的一部分,例如通过添加至靶标特异性引物的5'-末端。所述通用引物可以结合通用引发位点并指导从通用引发位点的引物延伸。
如本文所用,术语“靶序列”、“靶核酸”或“靶标”是指待检测或分析的样品中核酸序列的一部分。术语靶标包括靶序列的所有变体,例如,一种或多种突变型变体和野生型变体。
术语“测序”是指确定靶核酸中核苷酸序列的任何方法。
核酸测序正在被快速扩展至临床实践中。目前的测序技术采用单分子测序,并且允许检测极端罕见的靶标。例如,核酸测序已被用于检测脱落入患者血流的罕见肿瘤dna。检测单独分子通常需要分子条形码,诸如美国专利号7,393,665、8,168,385、8,481,292、8,685,678和8,722,368中所述。独特分子条形码是通常在体外操作的最早步骤期间添加至患者样品中的每个分子的短人工序列。所述条形码标记分子及其后代。所述独特分子条形码(uid)具有多种用途。条形码允许追踪样品中的每个单独的核酸分子,以评价例如患者血液中循环肿瘤dna(ctdna)分子的存在和量,以便在没有活检的情况下检测和监测癌症(newman,a.,等人,(2014)anultrasensitivemethodforquantitatingcirculatingtumordnawithbroadpatientcoverage,naturemedicinedoi:10.1038/nm.3519)。
独特分子条形码也可用于测序错误校正。单一靶分子的整个后代用相同的条形码标记并形成条形码化家族。没有被条形码化家族的所有成员共享的序列的变化作为伪像而非真正的突变被丢弃。条形码也可用于位置去重复(positionaldeduplication)和靶标定量,因为整个家族代表原始样品中的单一分子(newman,a.,等人,(2016)integrateddigitalerrorsuppressionforimproveddetectionofcirculatingtumordna,naturebiotechnology34:547)。
条形码实现的错误校正已大大增强测序测定的灵敏度。测序伪像诸如聚合酶错误不再是检测罕见点突变的障碍。同时,条形码对于检测易位(基因融合)(人恶性肿瘤中的另一种常见类型的突变)没有那么有益。参见f.mertens,等人.(2015)theemergingcomplexityofgenefusionsincancer,nat.rev.cancer15:371;f.mitelman,等人.(2007)theimpactoftranslocationsandgenefusionsoncancercausation,nat.rev.cancer7:233。由于条形码通常被随机连接至靶分子的两个末端,所以不知道哪些5'条形码最初与哪些3'条形码相关。这在文库制备的扩增步骤期间产生问题,因为嵌合分子经由pcr中的模板转换产生。存在于测序文库中的人工产生的嵌合分子与可能已存在于原始样品中的真实基因融合体不可区分。这直接限制检测低频基因融合体(这可以是癌症中重要的驱动突变)的能力。需要一种基于条形码的方法来追踪和消除人工基因融合体,以使得能够检测真正的突变。
在一些实施方案中,本发明是用于核酸测序的条形码化环状分子的文库。
在一些实施方案中,本发明是经由产生环状条形码化核酸分子的文库对核酸进行测序的方法。
在一些实施方案中,本发明是核酸测序中的错误校正方法,其利用条形码来鉴定原始样品中存在的基因融合分子。在该实施方案的变型中,本发明是核酸测序中的错误校正方法,其利用条形码来消除原始样品中不存在、但在核酸测序步骤期间产生的人工基因融合分子。
本发明包括通过核酸测序来检测样品中的靶核酸。可以使用本文所述的方法和组合物检测多种核酸,包括样品中的所有核酸。在一些实施方案中,所述样品来源于受试者或患者。在一些实施方案中,所述样品可以包含(例如通过活检)来源于受试者或患者的实体组织或实体瘤的片段。所述样品还可以包括体液(例如,尿液、痰液、血清、血浆或淋巴液、唾液、痰液、汗液、泪液、脑脊髓液、羊水、滑液、心包液、腹膜液、胸膜液、囊液、胆汁、胃液、肠液或粪便样品)。所述样品可以包括其中可存在正常或肿瘤细胞的全血或血液级分。在一些实施方案中,所述样品,尤其是液体样品,可以包含无细胞材料,诸如无细胞dna或rna,包括无细胞肿瘤dna或肿瘤rna。在一些实施方案中,所述样品是无细胞样品,例如无细胞血液来源的样品,其中存在无细胞肿瘤dna或肿瘤rna。在其他实施方案中,所述样品是培养的样品,例如含有或怀疑含有源自培养物中的细胞或源自培养物中存在的传染剂的核酸的培养物或培养物上清液。在一些实施方案中,所述传染剂是细菌、原生动物、病毒或支原体。
靶核酸是可以存在于样品中的目标核酸。在一些实施方案中,所述靶核酸是基因或基因片段。在一些实施方案中,所有基因、基因片段和基因间区域(整个基因组)构成靶核酸。在一些实施方案中,仅基因组的一部分,例如仅基因组的编码区(外显子组)构成靶核酸。在一些实施方案中,所述靶核酸含有遗传变体的基因座,例如,多态性,包括单核苷酸多态性或变体(snv的snp),或导致例如基因融合的遗传重排。在一些实施方案中,所述靶核酸包含生物标志物,即其变体与疾病或病况相关的基因。在其他实施方案中,所述靶核酸是特定生物体特征性的并且有助于鉴定生物体或病原生物体的特征,诸如药物敏感性或药物抗性。在还有其他实施方案中,所述靶核酸是人受试者特征性的,例如,定义受试者的独特hla或kir基因型的hla或kir序列。
在本发明的一个实施方案中,将一种或多种靶核酸转化为本发明的模板构型。在一些实施方案中,所述靶核酸在自然界中以单链形式(例如,rna,包括mrna、微小rna、病毒rna;或单链病毒dna)存在。在其他实施方案中,所述靶核酸在自然界中以双链形式存在。本领域技术人员将认识到,本发明的方法具有多个实施方案。可以将单链靶核酸转化为本发明的结构,如图1所示。可以将双链靶核酸转化为双链结构,其中每条链如图1中所描绘。或者,在遵循本文公开的方法的剩余步骤之前,可以首先将单链靶核酸转化为双链形式。可以将更长的靶核酸片段化,尽管在一些应用中可能需要更长的靶核酸以实现更长的读数。在一些实施方案中,所述靶核酸是天然片段化的,例如,循环的无细胞dna(cfdna)或化学降解的dna,诸如在保存的样品中发现的核酸。
本发明包括使用待连接至一个靶核酸的两个末端、因此形成环状分子的一个衔接子分子。在一些实施方案中,所述衔接子是与单链靶核酸分子连接的单链。在一些实施方案中,连接单链核酸使用夹板(splint)寡核苷酸进行,参见例如,美国申请公开号20120003657。在其他实施方案中,连接单链核酸或部分单链核酸使用5'-和3'-末端单链区域(突出端)进行,参见例如,美国申请公开号20140193860。在其他实施方案中,所述衔接子是与双链靶核酸分子连接的双链分子。双链分子的连接是本领域众所周知的(参见greenm.,和sambrook,j.,molecularcloning,2012cshlpress),并且本文描述了对一般方法的改进。在一些实施方案中,双链连接是平末端连接。在其他实施方案中,双链连接是t-a连接或其他突出端连接。在其他实施方案中,双链连接由拓扑异构酶驱动。
在一些实施方案中,双链衔接子在两个末端之一上具有光可裂解的间隔基。在该设置中,仅一个末端可以在连接反应中连接至文库分子。在连接时段后,将反应暴露于长波长uv(~350nm),裂解光可裂解的间隔基,并留下衔接子的磷酸化的5'-末端。连接反应可以继续形成环状模板。在一些实施方案中,在光裂解之后,稀释反应物以降低模板浓度并促进自连接成环。在该实施方案中,连接导致减少的伪像形成(例如,dna1-衔接子-dna2或衔接子1-dna-衔接子2)和更大的靶核酸分子(更大的ge(基因组当量))回收的回收。
在一些实施方案中,所述衔接子分子是体外合成的人工序列。在其他实施方案中,所述衔接子分子是体外合成的天然存在的序列。在还有其他实施方案中,所述衔接子分子是分离的天然存在的分子或分离的非天然存在的分子。
在一些实施方案中,所述衔接子包含一个或多个条形码。条形码可以是用于在样品被混合(多重化)的情况下标识样本来源的多重样品id(mid)。所述条形码还可以充当用于标识每个原始分子及其后代的独特分子id(uid)。所述条形码也可以是uid和mid的组合。在一些实施方案中,单个条形码用作uid和mid两者。
在一些实施方案中,每个条形码包含预定义序列。在其他实施方案中,所述条形码包含随机序列。条形码可以是1-20个核苷酸长。
在本发明的实施方案中,所述文库分子含有至少两个包括在衔接子中的条形码,所述衔接子与靶核酸连接。在本发明的一些实施方案中,所述条形码为约4-20个碱基长,使得96至384个不同的衔接子(每个衔接子具有不同对的相同条形码)被添加至人基因组样品。普通技术人员将认识到条形码的数目取决于样品的复杂性(即,独特靶分子的预期数目),并且能够为每个实验产生合适数目的条形码。
在一些实施方案中,本发明包括用于产生环状条形码化分子的文库的衔接子的合并物。合并物内的衔接子具有一对相同的条形码,其与合并物中的其他条形码相距至少1个或至少3个编辑距离。基于测序技术的典型错误率,本领域技术人员将能够确定哪个编辑距离对于特定实验是最佳的。通常,更大的编辑距离意味着可以在一个合并物中使用更少的条形码。然而,如果测序技术或制造过程具有高错误率,则将期望更大的编辑距离。例如,用于制备衔接子的寡核苷酸制造过程可具有高错误率。类似地,在边合成边测序工作流程中的dna扩增或引物延伸中使用的核酸聚合酶可以具有高错误率。这些错误率将需要增加合并物的衔接子中条形码间的编辑距离。相反,改进上面提及的每种方法的准确性将允许减少合并物的衔接子中的条形码间的编辑距离。
在一些实施方案中,本发明包括由含有整个衔接子的合并物的单个小瓶代表的制品。或者,制品可以包含试剂盒,其中合并物的一个或多个衔接子存在于分开的小瓶中。
所述衔接子进一步包含用于至少一种通用引物的引物结合位点。如果存在两个引物结合位点,则两个引物以相反的方向面向。本领域技术人员将认识到双链衔接子序列将在一条或两条链上具有引物结合位点。所述引物结合位点是与引物互补的序列,所述引物可以结合所述序列并促进链延伸。同时,本领域技术人员将认识到单链衔接子序列将具有用于第一引物的引物结合位点和与第二引物相同的序列。
在一些实施方案中,所述衔接子具有两个以相反方向面向的引物结合位点,以便使得能够复制每条链并随后pcr扩增两条链。在其他实施方案中,所述衔接子仅具有一个引物结合位点以使得能够仅复制一条链。在一些实施方案中,期望多于一轮复制。可以用相同的引物或不同的引物进行几轮。所述衔接子可以具有几个引物结合位点,例如第一引物结合位点内部的第二引物结合位点。或者,正向引物和反向引物中的一者或两者可以包含与所述衔接子不互补且包含额外引物结合位点的5'-襟翼序列。
在一些实施方案中,所述衔接子包含核酸合成终止(stop)位点。所述位点包含一个或多个核苷酸或核苷酸类似物,其不可被核酸聚合酶绕开。在一些实施方案中,所述stop位点是一个或多个核苷酸和核苷酸类似物,其选自无碱基核苷酸、具有蛋白侧基的核苷酸、合成核苷酸arac(阿糖胞苷)或脱氧尿嘧啶、异鸟嘌呤、5-甲基异胞嘧啶、乙二醇间隔基、具有大体积类似物诸如荧光团的核苷酸或非天然碱基对(ubp)“d5sics-dnam”核酸类似物(参见malyshev,d.,等人,(2012)efficientandsequence-independentreplicationofdnacontainingathirdbasepairestablishesafunctionalsix-lettergeneticalphabet.p.n.a.s.109(30):12005.)。本领域技术人员将理解终止子核苷酸可以对特定核酸聚合酶是特异性的,而其他核酸聚合酶能够绕过相同的终止子。例如,烷基化脱氧鸟嘌呤(n7和n2)通常是taqdna聚合酶的合成终止子。参见ponti,m.,等人(1991)measurementofthesequencespecificityofcovalentdnamodificationbyantineoplasticagentsusingtaqdnapolymerase,nucl.acidsres.19:2929。同样,dna中的脱氧尿嘧啶引起一些聚合酶的停止,而其他聚合酶绕过它。wardle,j.,等人(2008)uracilrecognitionbyreplicativednapolymerasesislimitedtothearchaea,notoccurringwithbacteriaandeukarya,nucl.acidsres.36(3):705-711。
在一些实施方案中,本发明利用酶。所述酶可以包括dna聚合酶(包括测序聚合酶)、dna连接酶和末端转移酶。
在一些实施方案中,所述dna聚合酶是高保真dna聚合酶,其在不寻常的碱基(即本发明中使用的stop位点)处有效终止合成。高保真聚合酶的实例是古细菌聚合酶,诸如pfu(来自激烈热球菌(pyrococcusfuriosus))。在其他实施方案中,使用taq聚合酶。在一些实施方案中,所述聚合酶具有3'-5'外切核酸酶活性。在其他实施方案中,所述聚合酶不具有链置换活性。
在一些实施方案中,本发明还利用dna连接酶。在一些实施方案中,使用t4dna连接酶或大肠杆菌dna连接酶。
在一些实施方案中,本发明还利用模板非依赖性dna聚合酶,例如末端转移酶或dna聚合酶,其具有以不依赖于模板的方式添加一个或多个核苷酸的活性。在一些实施方案中,本发明使用哺乳动物末端转移酶或taq聚合酶。
在一些实施方案中,本发明包括扩增步骤。该步骤可以涉及线性或指数扩增,例如pcr。扩增可以是等温的或涉及热循环。在一些实施方案中,所述扩增是指数的并涉及pcr。使用通用引物,即,单对引物与衔接子中的结合位点杂交。可以用相同的引物组扩增文库中的具有相同衔接子的所有分子。因为用通用引物的pcr具有降低的序列偏差,所以不需要限制扩增循环的数目。其中使用通用引物的扩增循环的数目可以是低的,但也可以是10个、20个或高达约30个或更多个循环,这取决于后续步骤所需的产物量。
测序
环状条形码化分子的文库和从文库生成的线性扩增子可以进行核酸测序。可以通过本领域已知的任何方法进行测序。特别有利的是高通量单分子测序。此类技术的实例包括illuminahiseq平台(illumina,sandiego,cal.)、iontorrent平台(lifetechnologies,grandisland,ny)、利用smrt的pacificbiosciences平台(pacificbiosciences,menlopark,cal.)或利用纳米孔技术的平台诸如由oxfordnanoporetechnologies(oxford,uk)或rochegenia(santaclara,cal.)制造的那些和任何其他目前存在或未来的涉及或不涉及边合成边测序的dna测序技术。测序步骤可以利用平台特异性测序引物。可以在扩增步骤中使用的扩增引物的5'-部分中引入这些引物的结合位点。如果条形码化分子的文库中不存在引物位点,则可以进行引入此类结合位点的额外的短扩增步骤。
在一些实施方案中,所述测序步骤涉及序列分析。在一些实施方案中,所述分析包括序列比对的步骤。在一些实施方案中,使用比对来确定来自多个序列(例如具有相同的条形码(uid)的多个序列)的共有序列。在一些实施方案中,使用条形码(uid)来确定来自都具有相同的条形码(uid)的多个序列的共有序列。在其他实施方案中,使用条形码(uid)来消除伪像,即,在一些但不是所有具有相同条形码(uid)的序列中存在的变异。可以消除由pcr错误或测序错误导致的此类伪像。
在一些实施方案中,可以通过定量样品中的具有每种条形码(uid)的序列的相对数目来定量样品中的每种序列的数目。每个uid代表原始样品中的单一分子,并且计数与每种序列变体相关的不同uid可以确定原始样品中每种序列的分数。本领域技术人员将能够确定测定共有序列所必需的序列读数的数目。在一些实施方案中,相关数目是精确定量结果所必需的每个uid的读数(“序列深度”)。在一些实施方案中,期望深度是每个uid5-50个读数。
在一些实施方案中,使用条形码(uid)来检测基因融合并消除模拟基因融合事件的伪像。在一些实施方案中,序列分析涉及将靶序列的读数与已知基因组序列比对的步骤。每个读数必须含有映射至目标基因组的靶序列和两个末端上的相同条形码(uid)。真正的基因融合分子将具有映射至靶基因组的不同区域的靶序列,但在两个末端上具有相同的条形码(uid)。具有映射至靶基因组的不同区域的靶序列、但在两个末端上具有不同的条形码的分子是伪像,而不是真正的基因融合分子。
本发明人已经观察到此类伪像以接近或超过体内存在的罕见基因融合分子的频率的频率存在。不受特定理论束缚,本发明人假设此类伪像在用文库分子进行pcr期间出现。通用引物的延伸可以在一个文库分子上开始并经历模板转换以在第二个文库分子上继续。所得融合分子将具有用于两个通用引物的结合位点,并在随后的pcr循环中扩增。使用根据本发明的条形码匹配从测序数据鉴定并消除此类分子。消除所述伪像允许以高得多的灵敏度和特异性检测真实的基因融合事件。
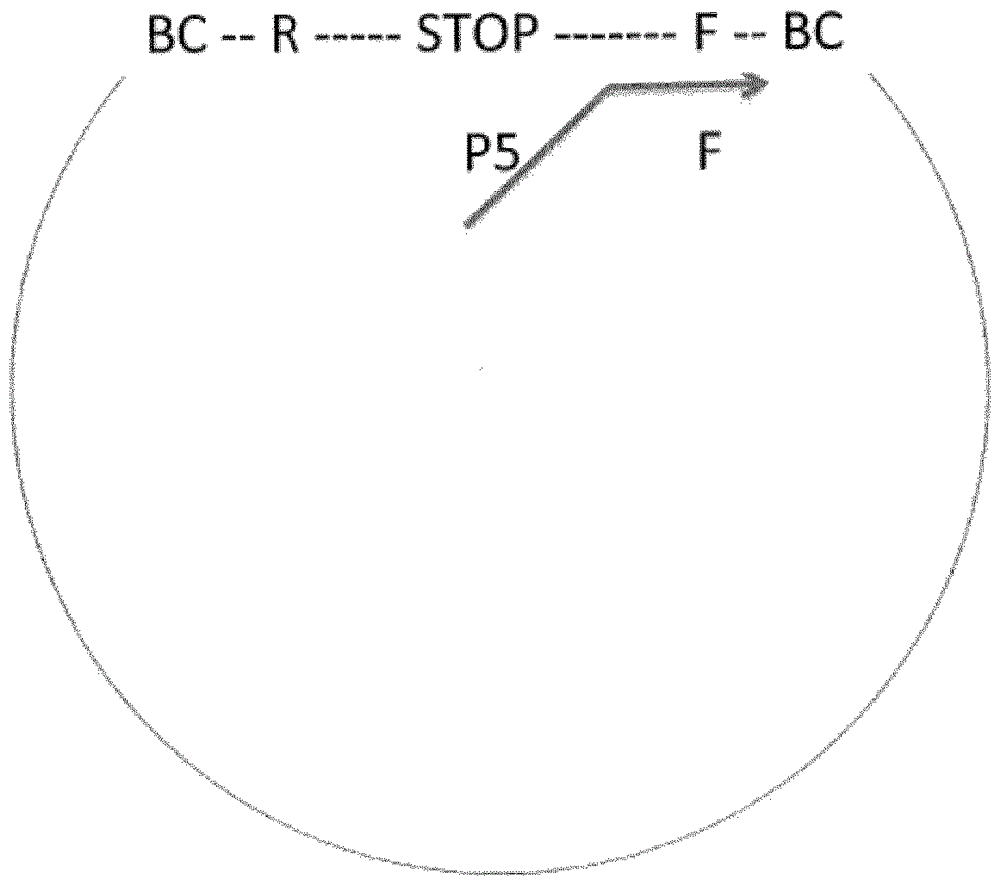
在图1-8中更详细地表示本发明。图1描绘根据本发明的单链(变性)文库分子。单个双链衔接子与双链靶分子的连接产生双链环状分子,其可以变性以产生图1中描绘的结构。bc是衔接子中存在的条形码。每个衔接子含有两个相同的条形码。在一些实施方案中,可以使用不同的条形码。在任一种情况下,条形码(或两个条形码的组合)在文库制备中使用的衔接子中是独特的。每个文库及其后代可以通过两个拷贝相同的独特条形码或两个条形码的独特组合独特地标识。r和f分别是反向和正向测序引物的结合位点。本领域技术人员将立即理解,核酸的单链(诸如图1中所描绘)含有一个引物(图1中的f引物)的结合位点(互补序列)和与相反面向的引物(图1中的r引物)相同的序列,而互补链(图1中未显示)将具有与f引物相同的序列和r引物的结合位点(互补序列)。stop是本文进一步描述的链合成终止子。
任选地,可以使用顺磁珠粒从样品分离图1中所示的环状模板。将与衔接子分子互补的不可延伸的捕获探针添加至样品中。可以使用两种捕获探针来捕获环状分子的每条链。捕获探针在3'-末端生物素化,并且可以用链霉抗生物素蛋白包被的顺磁珠粒捕获。所述探针可以具有以下结构:
图2描绘第一链合成的起始,其中f引物结合文库分子中的引物结合位点。引物在其5'末端处具有额外非互补序列。所述额外序列可以含有功能元件,例如测序引物结合位点(p5)。图3描绘第一链合成和stop处的终止。图4描绘图3的双链体环状分子和分离的(变性的)新合成的第一链,测序引物结合位点(p5)、正向引物(f)的序列,两个条形码(bc)侧接的靶序列和反向引物(r)的结合位点。在新合成的第一链中不存在stop。
图5描绘第一链合成的起始,其中r引物结合第一链中的引物结合位点。引物在其5'末端处具有额外非互补序列。所述额外序列可以含有功能元件,例如测序引物结合位点(p7)。图6描绘第二链合成,其复制第一链的所有元件,包括测序引物结合位点p5。图7描绘下一轮第一链合成的起始,其中p5测序引物结合第二链中的其结合位点。
图8描绘最终的线性双链文库分子,其准备用于进一步步骤,诸如扩增和测序。双链分子含有测序(或扩增)引物结合位点p5和p7以及条形码(bc)。该分子还保留初始的正向和反向引物结合位点f和r。双链文库分子的特征在于独特条形码(或条形码的独特组合),其将该分子及其后代与样品中的所有其他分子及其后代区分。
实施例1(预言性)
产生条形码化环状分子的文库
在本实施例中,从样品分离dna。将分离的dna任选地片段化并针对环状分子的最佳尺寸进行尺寸选择。在存在罕见靶核酸的情况下,可以省略尺寸选择步骤。在一些情况下,从样品分离rna并逆转录成cdna,并在随后的步骤中如同从样品直接分离的dna一样进行处理。
将dna用t4dna聚合酶进行末端修复和加a-尾。a-尾的添加允许随后的有效连接,避免来自平端连接的复杂化。
接下来,将双链接头连接至输入dna以形成环状分子。所述双链接头具有以下结构
其中,5'p是5'-磷酸酯,[t]是添加的t,其与靶分子的3'-末端处的a碱基配对,bc是条形码,stop是终止子核苷酸,且r和f分别是反向和正向引物结合位点。任选地,用t7外切核酸酶处理样品以除去未环化的dna和过量的衔接子(样品中剩余的具有游离末端的任何dna)。
任选地,可以使用顺磁珠粒从样品分离环状模板。使用两种不可延伸的捕获探针来捕获环状分子的每条链。捕获探针在3'-末端处生物素化,并且用链霉抗生物素蛋白包被的顺磁珠粒捕获。所述捕获过程包括以下步骤:热变性,与珠粒结合,使用磁体从溶液移取珠粒捕获的dna,任选地洗涤珠粒,以及通过在升高温度下变性从珠粒结合的捕获探针洗脱。
然后通过pcr扩增分离的环状模板。用与衔接子中的引物结合位点互补的引物进行pcr。每个引物具有与衔接子中的结合位点不互补且含有测序引物结合位点或流动室结合序列(这取决于测序仪器和技术的选择)的5'-襟翼。pcr产生线性分子。环状模板分子的每条链中的stop碱基阻止聚合酶完成环。
接下来,分析从线性模板导出的序列数据。具有映射至靶基因组的不同区域的靶序列、但在两个末端上具有相同的(或先前匹配的)条形码的分子被检测为真正的基因融合分子。具有映射至靶基因组的不同区域的靶序列、但在两个末端上具有不同的(或先前未匹配的)条形码的分子是从测序数据丢弃的伪像。
- 还没有人留言评论。精彩留言会获得点赞!