1,3-二芳基-1,5,6,7-四氢吲唑衍生物的合成方法与流程
本发明涉及吲唑类化合物的合成
技术领域:
。
背景技术:
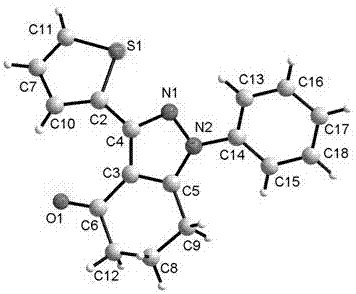
:吲唑类化合物具有重要的生物活性,已广泛用于临床治疗如作为酪氨酸激酶(VEGFR)小分子抑制剂GW-786034(pazopanib)具有很好的抗肿瘤和抑制血管生成的活性,用于治疗肾癌、乳腺癌、卵巢癌等。AG-013736(axitinib)可用于治疗NSCLC、甲状腺癌及黑色素瘤等[茆勇军,田广辉,王震中国新药杂志,2008,17,544.]。氯尼达明(Doridamina)(化学简称LND)已成为一种口服抗癌药物[唐家邓,岑均达中国医药工业杂志,2009,40,12;侯长军,罗斌,霍丹群药学进展,2006,30,235.]。吲唑类化合物也用作肿瘤放疗靶向性增敏剂药物[王浩靶向肿瘤放疗增敏剂-NIES系列化合物的研究北京:中国医学科学院,2009.]。研究表明吲唑类衍生物可以作为人类免疫缺陷病毒蛋白酶抑制剂(HIVPRI)和非核普类逆转录酶抑制剂(NNRTI),其对逆转录酶在病毒感染的细胞内的存活周期有很重要的影响,能够有效治疗艾滋病[Sun,J.H.;Teleha,C.A.;Yan,J.S.J.Org.Chem.1997,62,5627;Jones,L.H..;Allan,G.;Barba,O.J.Med.Chem.2009,52,1219.]。吲唑衍生物作为5-HT2c受体激动剂,在治疗焦虑、癫痫、肥胖症和强迫症等疾病方面有一定的疗效[沈晓红5-HT2c受体激动剂小分子化合物的合成上海:华东师范大学,2006.]。含吲唑单元的苄达酸(Bendazac)临床用作消炎镇痛药[金初瑢,魏传喜华西药学杂志,1990,5,32.]。含吲唑单元的格拉司琼(Granisetron),一种具有高度选择性的5-HT3受体拮抗剂,对预防和治疗放化疗过程中产生的恶心、呕吐不良反应,有较好的疗效[Vernekar,S.K.V.;Hallaq,H.Y.;Clarkson,G.J.Med.Chem.2010,53,2324.]。含吲唑单元的化合物也常用于预防和治疗疟疾[德奥池蒙特H,弗赖塞L,兹默曼ACN1933834A,2007.]。而1-芳基吲唑类衍生物通常可用作避孕药[Lebedev,A.Y.;Khartulyari,A.S.;Voskoboynikov,A.Z.J.Org.Chem.2005,70,596.]。2-苄基-3-芳基-7-三氟甲基吲唑衍生物可用作肝X受体(LXRs)治疗动脉粥样硬化、Ⅱ型糖尿病等[Jay,W.;Robert,S.S.;Mare,B.J.Med.Chem.2008,51,7161.]。此外,吲唑衍生物可作为农药,如吲熟酯(Figaro)作为植物生长调节剂,具有催熟作用,可用于苹果、相桔、梨等果树果实的催熟。同时,它还具有促进果实增厚,增加果肉的糖和氨基酸含量,防止果皮破裂等作用,可以提高果实的品质[南开大学元素有机化学教研所国外农药进展(三)北京:化学工业出版社,1992.]。如四氢吲唑衍生物对杀除水稻杂草具有理想的活性和选择性[Hwang,I.T.;Kim,H.R.;Jeon,D.J.;Hong,K.S.;Song,J.H.;Chung,C.K.;Cho,K.Y.PestManag.Sci.2005,61,483.]。吲唑衍生物中四氢吲唑类化合物往往具有特殊的生物活性,如6,6-二甲基-1,3-二取代四氢吲唑是一种非肽类鸦片受体激动剂可用作兴奋剂[Cheng,M.F.;Ou,L.C.;Chen,S.C.;Chang,W.T.;Law,P.Y.;Loh,H.H.;Chao,Y.S.;Shih,C.;Yeh,S.H.;Ueng,S.H.Bioorg.Med.Chem.2014,22,4694.]。4,5-二氢吲唑衍生物对大麻素有着良好的拮抗作用,可用作减肥药[Lazzari,P.;Loriga,G.;Manca,I.;Pinna,G.A.;Pani,L.US20100216785A1,2010.]。许多四氢吲唑化合物可以作为热休克蛋白90抑制剂而影响肿瘤细胞的生长和基因转录,用于特效抗肿瘤试剂[Khlebnicova,T.S.;Piven,Y.A.;Baranovsky,A.V.;Lakhvich,F.A.;Shishkina,S.V.;Zicane,D.;Tetere,Z.;Ravina,I.;Kumpinš,V.;Rijkure,I.;Mierina,I.;Peipinš,U.;Turks,M.Steroids.2017,117,77.]。由于吲唑衍生物重要的生理活性及潜在的药用价值,对它们合成方法的研究一直备受关注。已报道了一系列合成方法,如2013年,Barraja等人[Barraja,P.;Spano,V.;Giallombardo,D.;Diana,P.;Montalbano,A.;Carbone,A.;Parrino,B.;Cirrincione,G.Tetrahedron2013,69,6474.]通过分步合成的方法先将1,3-环己二酮与肼类合成腙类化合物,然后与醛在哌啶/醋酸催化下得到2,3-二取代四氢吲唑类化合物。2001年,Yen等人[Yen,Y.P.;Chen,S.F.;Heng,Z.C.;Huang,J.C.;Kao,L.C.;Lai,C.C.;Liu,R.S.H.Heterocycles2001,55,1859.]使用螺-3H-吡唑类化合物经过两步加热的方法最终扩环得到1,3-二取代四氢吲唑类化合物。2011年,James等人[Counceller,C.M.;Eichman,C.C.;Wray,B.C.;Welin,E.R.;Stambuli,J.P.Org.Synth.2011,88,33.]将邻氨基苯乙酮先制成羟胺化合物,然后在NEt3碱性条件以及MsCl催化下直接分子内环合得到3-甲基吲唑。1990年,Takefumi等人[Momose,T.;Toyooka,N.;Ikuta,T.;Yanagino,H.Heterocycles1990,30,789.]先将N-不饱和环基甲基酰胺与N2O4,醋酸钾一起在乙二醇二甲醚中-10oC反应得到氧代酰肼化合物,再与碳酸钙在二氧六环中加热85-110oC最终得到吡唑稠环类化合物。2014年,Houk等人[Medina,J.M.;McMahon,T.C.;Jimenez-Oses,G.;Houk,K.N.;Garg,N.K.J.Am.Chem.Soc.2014,136,14706.]发现了将α,β-不饱和三氟苯磺基三甲基硅基环己(戊)烯与引发剂在CsF催化下反应能够成功合成出来多种吡唑稠环类衍生物。2013年,Lee等人[Lee,H.K.;Cho,C.S.Synth.Commun.2013,43,915.]通过将取代1-(2-溴苯(或环己基)亚甲基)-2-取代肼在铜粉和叔丁醇钠的协调促进下进行分子内偶联获得多取代吲唑(或四氢吲唑)衍生物。1999年,Katritzky等人[Katritzky,A.R.;Denisenko,A.;Denisenko,S,N.J.HeterocyclicChem.2000,37,1309.]以苯并三氮唑-1-甲醛二甲亚胺盐与环己酮亚胺为底物分两步反应最终得到四氢吲唑及其衍生物。1978年,Nagakura等人[Nagakura,M.;Ota,T.;Shimidzu,N.;Kawamura,K.;Eto,Y.;Wada,Y.J.Med.Chem.1979,22,48.]将取代2-亚甲基羟基环己酮与取代肼在甲醇中45-50oC加热反应6h最终得到1-取代的四氢吲唑和2-取代的四氢吲唑。2017年,Hamama等人[Hamama,W.S.;El-Din,A.;Hassanien,E.;Zoorob,H.H.J.HeterocyclicChem.2017,doi:10.1002/jhet.2947.]利用3-((二甲胺基)亚甲基)萘-1,2,4(3H)-四酮与取代肼在DMF下回流6-8h最终获得2-取代吡唑稠环类衍生物。2017年,Wu等人[Yu,Y.;Chen,Y.;Huang,W.;Wu,W.Q.;Jiang,H.F.J.Org.Chem.2017,82,9479.]先将环酮类化合物与对甲苯磺酰肼在乙醇中70℃反应2h得到腙类化合物,然后将其与碳化钙,碳酸铯混合,改用水和DMSO作溶剂,80oC反应6h,最终得到多种吡唑稠环类化合物。除了传统的环化合成途径,已有吡唑稠环的官能团化是拓展其衍生物的一个重要途径,如2013年,Yu等人[Ye,M.;Edmunds,A.J.F.;Morris,J.A.;Sale,D.;Zhanga,Y.;Yu,J.Q.Chem.Sci.2013,4,2374.]通过钯催化成功将芳基偶联接入(四氢)吲唑的C(3)位得到一系列衍生物。目前文献方法往往存在合成路线长,总收率低,使用不易获得的三羰基原料,底物肼和二羰基缩合成环缺少区域选择性而生成分离困难的同分异构体,或者使用贵金属催化剂等缺点。技术实现要素:针对现有技术合成存在的以上缺陷,本发明提出一种简单、具有高度区域选择性的1,3-二芳基-1,5,6,7-四氢吲唑衍生物的合成方法。本发明技术方案是:以三乙烯二胺为催化剂,在溶剂存在下,将取代1,3-环己二酮、取代β-硝基芳基乙烯和取代苯肼混合进行反应,反应结束后减压蒸除残留溶剂后加水,取得有机相依次以水、饱和食盐水洗涤后,再经干燥,取得粗产品通过柱色谱分离,得1,3-二芳基-1,5,6,7-四氢吲唑衍生物。反应式如下:上式中,R1为H或5,5-diMe;R2为H、4-Me或4-Cl;Ar为Ph、4-MeC6H4、4-MeOC6H4、4-ClC6H4、4-BrC6H4、4-NO2C6H4、2-MeC6H4、3-MeC6H4、3-MeOC6H4、Furan-2-、Thiophen-2-、Thiophen-3-或3,4-OCH2CH2OC6H3。本发明以取代β-硝基芳基乙烯、取代1,3-环己二酮和取代苯肼为基本底物,在催化剂的作用下进行反应,提供了一种简单的由β-硝基芳基乙烯,1,3-环己二酮和苯肼多组分一锅煮反应合成1,3-二芳基-1,5,6,7-四氢吲唑衍生物的方法。本发明使用一锅煮反应合成目标化合物,收率高,原料易得,尤其底物肼参与反应具有高度的区域选择性,产物单一,后处理简洁。此外,本方法无需使用贵金属催化剂等。进一步地,本发明先将取代1,3-环己二酮、取代β-硝基芳基乙烯、三乙烯二胺和溶剂混合后,再加入取代苯肼。本发明使用三乙烯二胺作为促进剂,首先促进取代1,3-环己二酮和取代β-硝基芳基乙烯的Micheal加成反应,在取代1,3-环己二酮的C2上引入具有导向基团的芳亚甲基,待反应基本完成后,随后加入取代苯肼,苯肼的空间位阻小的NH2能够选择性的进攻取代1,3-环己二酮的C2上的亚甲基,最后苯肼的空间位阻稍大的NH进攻取代1,3-环己二酮的酮羰基成环,获得了立体专一的产物。经试验表明取代1,3-环己二酮取代、β-硝基芳基乙烯和取代苯肼的投料摩尔比从1∶1∶1到1.2∶1∶1.05都能够以相应收率获得目标化合物。所述取代1,3-环己二酮、取代β-硝基芳基乙烯和取代苯肼的最佳投料摩尔比为1.1∶1∶1.05,收率可达83%。经试验表明取代β-硝基芳基乙烯和催化剂三乙烯二胺(DABCO)的投料摩尔比对反应影响比较大,使用为0.25∶1摩尔比,目标化合物的收率为43%,很显然三乙烯二胺(DABCO)的使用量不能够促进反应充分进行,而0.5∶1摩尔比能够有效地促进反应,而三乙烯二胺(DABCO)和β-硝基芳基乙烯的投料摩尔比为0.75∶1已达到最佳。为提高产率,通过改变反应温度研究对反应的影响,发现升高温度至50℃、60℃产率较大,可达83%,继续提高反应温度产率反而开始下降,由此确定了此条件下最佳反应温度为50~60℃。不同反应溶剂对反应影响较大,乙腈和甲醇有利于目标化合物的形成,而极性比较大的N,N-二甲基甲酰胺不利于目标化合物的形成。而二氯甲烷和甲苯作为在反应温度为50℃的条件下,反应难以有效发生。而采用乙腈为溶剂收率可达83%。附图说明图1为1-苯基-3-对甲苯基-1,5,6,7-4H-四氢吲唑-4-酮(4b)的分子结构图。图2为1-苯基-3-(2-噻唑基)-1,5,6,7-4H-四氢吲唑-4-酮(4p)的分子结构图。具体实施方式一、1,3-二芳基-1,5,6,7-四氢吲唑衍生物的合成:将1.1mmol取代环己二酮(1a-b)、1.0mmol取代β-硝基芳基乙烯(2a-m)和0.75mmol三乙烯二胺(DABCO)投入6mL乙腈溶液中,常温下搅拌30分钟后,室温下向混合体系中投入1.05mmol取代苯肼(3a-c),升温至50℃继续搅拌8~12h,TLC薄层层析法(展开剂由体积比为1:3的乙酸乙酯和石油醚混合组成)跟踪至反应结束。减压蒸除残留溶剂后加入15mL水,搅拌后静置分层,分出有机相,并将水相用二氯甲烷萃取两次,每次二氯甲烷的用量为15mL。合并有机相和二氯甲烷萃取液。将合并的有机相依次用10mL水、15mL饱和食盐水洗涤一遍后,再以无水硫酸钠干燥,取得粗产品通过柱色谱(硅胶,EA:PE=1:5)分离,得到目标产物(4a-r)。本发明反应式如下:上式中,R1为H或5,5-diMe;R2为H、4-Me或4-Cl;Ar为Ph、4-MeC6H4、4-MeOC6H4、4-ClC6H4、4-BrC6H4、4-NO2C6H4、2-MeC6H4、3-MeC6H4、3-MeOC6H4、Furan-2-、Thiophen-2-、Thiophen-3-或3,4-OCH2CH2OC6H3。表1为1,3-二芳基-1,5,6,7-四氢吲唑衍生物的合成结果对比表:序号R1ArR2产物收率(%)1HPhH4a832H4-MeC6H4H4b823H4-MeOC6H4H4c884H4-ClC6H4H4d735H4-BrC6H4H4e676H4-NO2C6H4H4f527H2-MeC6H4H4g628H3-MeC6H4H4h699H3-MeOC6H4H4i74105,5-diMePhH4j84115,5-diMe4-MeOC6H4H4k91125,5-diMe4-ClC6H4H4l7513H4-ClC6H44-Me4m7814H4-ClC6H44-Cl4n5115HFuran-2-H4o6916HThiophen-2-H4p6417HThiophen-3-H4q6318H3,4-OCH2CH2OC6H3H4r45二、将以上18种产物结构通过核磁共振氢谱、碳谱、红外光谱、高分辨质谱等进行了表征,并通过单晶X-衍射的分析结果确定了它们的立体化学结构,各分子结构式及实验数据如下:(1)1,3-二苯基-1,5,6,7-4H-四氢吲唑-4-酮(4a):分子结构式为:实验数据:黄色固体;收率:83%;m.p.145.1-145.3°C(PE/EA);1HNMR(CDCl3,400MHz)δ(ppm):8.29(s,1H),8.04(d,J=7.2Hz,1H),7.50-7.44(m,4H),7.38-7.36(m,3H),7.31(dd,J=6.8,6.8Hz,1H),2.92(t,J=6.0Hz,2H),2.53(t,J=6.0Hz,2H),2.13-2.07(m,2H);13CNMR(CDCl3,150MHz)δ(ppm):192.8,151.9,150.9,138.6,131.9,129.4,128.9,128.9,128.3,128.0,124.1,116.8,39.1,23.8,23.3;IR(KBr,cm-1):ν3029,2946,1660,1599,1501,1449,1169,1025,911,767;HRMS(ESI)m/z[M+H]+calcdforC19H16N2O:289.1341;found:289.1344。(2)1-苯基-3-对甲苯基-1,5,6,7-4H-四氢吲唑-4-酮(4b):分子结构式:实验数据:黄色固体;收率:82%;m.p.164.6-165.2°C(PE/EA);1HNMR(CDCl3,400MHz)δ(ppm):8.01(d,J=8.0Hz,2H),7.57-7.50(m,4H),7.43(dd,J=7.2,6.8Hz,1H),7.24(d,J=8.0Hz,2H),2.99(t,J=6.0Hz,2H),2.60(t,J=6.0Hz,2H),2.39(s,3H),2.21-2.14(m,2H);13CNMR(CDCl3,100MHz)δ(ppm):192.6,151.9,150.7,138.7,138.6,129.3,128.9,128.8,128.7,128.2,124.1,116.7,39.0,23.8,23.3,21.3;IR(KBr,cm-1):ν3030,2944,1662,1599,1500,1448,1171,1026,913,831,765;HRMS(ESI)m/z[M+H]+calcdforC20H18N2O:303.1497;found:303.1497。(3)1-苯基-3-对甲氧苯基-1,5,6,7-4H-四氢吲唑-4-酮(4c):分子结构式:实验数据:黄色固体;收率:88%;m.p.182.7-183.6°C(PE/EA);1HNMR(CDCl3,400MHz)δ(ppm):8.12(d,J=8.8Hz,2H),7.57-7.50(m,4H),7.43(dd,J=7.2,6.8Hz,1H),6.96(d,J=8.8Hz,2H),3.85(s,3H),2.99(t,J=6.0Hz,2H),2.61(t,J=6.0Hz,2H),2.21-2.15(m,2H);13CNMR(CDCl3,100MHz)δ(ppm):192.7,160.1,151.6,150.7,138.6,130.3,129.3,128.2,124.4,124.1,116.5,113.4,55.2,39.1,23.8,23.3;IR(KBr,cm-1):ν3033,2941,1661,1593,1499,1450,1182,1027,910,828,762;HRMS(ESI)m/z[M+H]+calcdforC20H18N2O2:319.1447;found:319.1450。(4)1-苯基-3-对氯苯基-1,5,6,7-4H-四氢吲唑-4-酮(4d):分子结构式:实验数据:黄色固体;收率:73%;m.p.147.2-147.7°C(PE/EA);1HNMR(CDCl3,400MHz)δ(ppm):8.12(d,J=8.4Hz,2H),7.57-7.51(m,4H),7.45(dd,J=6.8,7.2Hz,1H),7.40(d,J=8.4Hz,2H),3.00(t,J=6.0Hz,2H),2.62(t,J=6.0Hz,2H),2.22-2.15(m,2H);13CNMR(CDCl3,100MHz)δ(ppm):192.7,151.0,150.7,138.4,134.7,130.3,130.2,129.4,128.4,128.2,124.1,116.7,39.0,23.7,23.2;IR(KBr,cm-1):ν3031,2943,1660,1595,1498,1451,1188,1029,913,826,764;HRMS(ESI)m/z[M+H]+calcdforC19H15ClN2O:323.0951;found:323.0958。(5)1-苯基-3-对溴苯基-1,5,6,7-4H-四氢吲唑-4-酮(4e)分子结构式:实验数据:黄色固体;收率:67%;m.p.189.6-190.1°C(PE/EA);1HNMR(CDCl3,400MHz)δ(ppm):8.06(d,J=8.4Hz,2H),7.56-7.51(m,6H),7.45(dd,J=6.4,6.4Hz,1H),7.40(d,J=8.4Hz,2H),2.99(t,J=6.0Hz,2H),2.61(t,J=6.0Hz,2H),2.21-2.15(m,2H);13CNMR(CDCl3,100MHz)δ(ppm):192.7,151.0,150.7,138.4,131.1,130.8,130.5,129.4,128.4,124.1,123.1,116.7,39.0,23.7,23.2;IR(KBr,cm-1):ν3032,2941,1661,1599,1501,1449,1188,1026,917,829,762;HRMS(ESI)m/z[M+H]+calcdforC19H15BrN2O:367.0446;found:367.0447。(6)1-苯基-3-对硝基苯基-1,5,6,7-4H-四氢吲唑-4-酮(4f):分子结构式:实验数据:黄色固体;收率:52%;m.p.177.1-177.4°C(PE/EA);1HNMR(CDCl3,400MHz)δ(ppm):8.40(d,J=8.8Hz,2H),8.28(d,J=8.8Hz,2H),7.57-7.56(m,4H),7.52-7.48(m,1H),3.03(t,J=6.0Hz,2H),2.66(t,J=6.0Hz,2H),2.25-2.19(m,2H);13CNMR(CDCl3,150MHz)δ(ppm):192.8,151.5,149.5,147.8,138.3,138.3,129.7,129.5,128.8,124.2,123.3,117.2,39.0,23.8,23.3;IR(KBr,cm-1):ν3034,2944,1661,1596,1499,1451,1187,1027,913,825,763;HRMS(ESI)m/z[M+H]+calcdforC19H15BrN2O:367.0446;found:367.0447。(7)1-苯基-3-邻甲苯基-1,5,6,7-4H-四氢吲唑-4-酮(4g):分子结构式:实验数据:黄色固体;收率:62%;m.p.148.6-149.3°C(PE/EA);1HNMR(CDCl3,400MHz)δ(ppm):7.59-7.57(m,2H),7.51(dd,J=7.6,8.0Hz,2H),7.42(d,J=7.6Hz,2H),7.32-7.21(m,3H),3.05(t,J=6.0Hz,2H),2.55(t,J=6.0Hz,2H),2.33(s,3H),2.23-2.20(m,2H);13CNMR(CDCl3,100MHz)δ(ppm):192.4,151.7,149.6,138.6,137.3,131.8,130.2,130.0,129.3,128.5,128.0,125.2,123.7,118.1,38.6,23.8,23.5,20.0;IR(KBr,cm-1):ν3022,2952,1661,1598,1500,1448,1167,1025,910,768;HRMS(ESI)m/z[M+H]+calcdforC20H18N2O:303.1497;found:303.1495。(8)1-苯基-3-间甲苯基-1,5,6,7-4H-四氢吲唑-4-酮(4h):分子结构式:实验数据:黄色固体;收率:69%;m.p.160.4-161.1°C(PE/EA);1HNMR(CDCl3,400MHz)δ(ppm):7.92(d,J=8.8Hz,1H),7.91(s,1H),7.58-7.51(m,4H),7.44(dd,J=6.4,7.2Hz,1H),7.33(dd,J=7.6,7.2Hz,1H),7.21(d,J=7.6Hz,1H),3.00(t,J=6.0Hz,2H),2.61(t,J=6.0Hz,2H),2.41(s,3H),2.22-2.15(m,2H);13CNMR(CDCl3,100MHz)δ(ppm):192.6,152.0,150.7,138.6,137.5,131.6,129.6,129.3,129.3,128.3,127.9,126.2,124.1,116.7,39.0,23.8,23.3,21.4;IR(KBr,cm-1):ν3028,2933,1667,1598,1500,1463,1221,1036,961,765,721;HRMS(ESI)m/z[M+H]+calcdforC20H18N2O:303.1497;found:303.1489。(9)1-苯基-3-间甲氧苯基-1,5,6,7-4H-四氢吲唑-4-酮(4i):分子结构式:实验数据:黄色固体;收率:74%;m.p.178.8-179.5°C(PE/EA);1HNMR(CDCl3,400MHz)δ(ppm):7.75(s,1H),7.65(d,J=7.6Hz,1H),7.49-7.42(m,4H),7.36(dd,J=7.2,7.2Hz,1H),7.26(dd,J=8.0,7.6Hz,1H),6.87(dd,J=8.0,0.8Hz,1H),3.80(s,3H),2.91(t,J=6.0Hz,2H),2.53(t,J=6.0Hz,2H),2.12-2.06(m,2H);13CNMR(CDCl3,100MHz)δ(ppm):192.7,159.3,151.7,150.9,138.5,133.1,129.3,128.9,128.3,124.1,121.2,115.5,113.8,108.6,55.3,39.1,23.8,23.2;IR(KBr,cm-1):ν3034,2924,1662,1595,1500,1460,1222,1034,965,759,722;HRMS(ESI)m/z[M+H]+calcdforC20H18N2O2:319.1447;found:319.1442。(10)6,6-二甲基-1,3-二苯基-1,5,6,7-4H-四氢吲唑-4-酮(4j):分子结构式:实验数据:黄色固体;收率:84%;m.p.120.6-121.0°C(PE/EA);1HNMR(CDCl3,400MHz)δ(ppm):8.15(d,J=6.8Hz,2H),7.58-7.52(m,4H),7.47-7.39(m,4H),2.85(s,2H),2.50(s,2H),1.14(s,6H);13CNMR(CDCl3,100MHz)δ(ppm):192.2,151.6,150.0,138.6,131.7,129.3,128.8,128.8,128.3,128.0,124.2,53.2,37.4,35.3,29.6,28.2;IR(KBr,cm-1):ν3024,2953,1662,1599,1500,1442,1178,1023,936,762;HRMS(ESI)m/z[M+H]+calcdforC21H20N2O:317.1654;found:317.1655。(11)6,6-二甲基-1-苯基-3-对甲氧苯基-1,5,6,7-4H-四氢吲唑-4-酮(4k):分子结构式:实验数据:黄色固体;收率:91%;m.p.138.2-138.9°C(PE/EA);1HNMR(CDCl3,400MHz)δ(ppm):8.16(d,J=8.4Hz,2H),7.59-7.51(m,5H),7.44(dd,J=7.2,6.8Hz,1H),6.96(d,J=8.8Hz,2H),3.85(s,3H),2.83(s,2H),2.49(s,2H),1.13(s,6H);13CNMR(CDCl3,100MHz)δ(ppm):192.2,160.2,151.3,150.0,138.6,130.2,129.3,128.2,124.3,124.2,113.4,55.2,55.2,53.2,37.4,35.2,28.2;IR(KBr,cm-1):ν3028,2954,1663,1598,1500,1449,1167,1027,926,830,765;HRMS(ESI)m/z[M+H]+calcdforC22H22N2O2:347.1760;found:347.1763。(12)6,6-二甲基-1-苯基-3-对氯苯基-1,5,6,7-4H-四氢吲唑-4-酮(4l):分子结构式:实验数据:黄色固体;收率:75%;m.p.123.4-123.8°C(PE/EA);1HNMR(CDCl3,400MHz)δ(ppm):8.16(d,J=8.8Hz,2H),7.55(d,J=8.4Hz,4H),7.49-7.44(m,1H),7.70(d,J=8.8Hz,2H),2.84(s,2H),2.50(s,2H),1.14(s,6H);13CNMR(CDCl3,100MHz)δ(ppm):192.2,150.4,150.2,138.4,134.7,130.3,130.2,129.4,128.5,128.2,124.2,53.1,37.4,35.3,29.6,28.2;IR(KBr,cm-1):ν3043,2959,1661,1601,1500,1453,1174,1028,928,828,765;HRMS(ESI)m/z[M+H]+calcdforC21H19ClN2O:351.1264;found:351.1261。(13)1-对甲苯基-3-对氯苯基-1,5,6,7-4H-四氢吲唑-4-酮(4m):分子结构式:实验数据:黄色固体;收率:78%;m.p.143.2-144.0°C(PE/EA);1HNMR(CDCl3,400MHz)δ(ppm):8.12(d,J=8.4Hz,2H),7.42(d,J=8.4Hz,2H),7.39(d,J=8.4Hz,2H),7.32(d,J=8.4Hz,2H),2.96(t,J=6.0Hz,2H),2.60(t,J=6.0Hz,2H),2.43(s,3H),2.20-2.14(m,2H);13CNMR(CDCl3,150MHz)δ(ppm):192.9,151.0,150.5,138.6,136.0,134.7,130.4,130.3,129.9,128.2,124.0,116.6,39.1,23.7,23.3,21.2;IR(KBr,cm-1):ν3044,2959,1682,1601,1506,1455,1173,1029,933,825,766;HRMS(ESI)m/z[M+H]+calcdforC20H17ClN2O:337.1108;found:337.1109。(14)1,3-二(对氯苯基)-1,5,6,7-4H-四氢吲唑-4-酮(4n):分子结构式:实验数据:黄色固体;收率:51%;m.p.158.9-159.7°C(PE/EA);1HNMR(CDCl3,400MHz)δ(ppm):8.02(d,J=8.8Hz,2H),7.43(s,4H),7.32(d,J=8.4Hz,2H),2.91(t,J=6.0Hz,2H),2.54(t,J=6.0Hz,2H),2.15-2.09(m,2H);13CNMR(CDCl3,100MHz)δ(ppm):192.7,151.0,151.0,137.0,134.9,134.3,130.3,130.1,129.6,128.3,125.2,117.0,39.0,23.8,23.3;IR(KBr,cm-1):ν3048,2952,1681,1600,1497,1451,1175,1023,935,822,766;HRMS(ESI)m/z[M+H]+calcdforC19H14Cl2N2O:357.0561;found:357.0561。(15)1-苯基-3-(2-呋喃基)-1,5,6,7-4H-四氢吲唑-4-酮(4o):分子结构式:实验数据:黄色固体;收率:69%;m.p.142.5-142.9°C(PE/EA);1HNMR(CDCl3,400MHz)δ(ppm):7.89(d,J=2.4Hz,1H),7.57-7.50(m,5H),7.44(dd,J=7.2,2.8Hz,1H),6.53(d,J=2.0Hz,1H),2.98(t,J=6.0Hz,2H),2.63(t,J=6.0Hz,2H),2.21-2.17(m,2H);13CNMR(CDCl3,100MHz)δ(ppm):192.2,150.4,146.6,143.0,142.5,138.3,129.3,128.4,124.3,115.8,113.8,111.4,38.7,23.5,23.2;IR(KBr,cm-1):ν3048,2953,1692,1663,1599,1489,1447,1385,1172,1022,913,768;HRMS(ESI)m/z[M+H]+calcdforC17H14N2O2:279.1134;found:279.1138。(16)1-苯基-3-(2-噻唑基)-1,5,6,7-4H-四氢吲唑-4-酮(4p):分子结构式:实验数据:黄色固体;收率:64%;m.p.149.8-150.3°C(PE/EA);1HNMR(CDCl3,400MHz)δ(ppm):8.50(d,J=2.8Hz,1H),7.56-7.50(m,4H),7.43(dd,J=7.2,6.8Hz,1H),7.33(d,J=4.8Hz,1H),7.12(dd,J=3.6,4.8Hz,1H),2.98(t,J=6.0Hz,2H),2.63(t,J=6.0Hz,2H),2.20-2.14(m,2H);13CNMR(CDCl3,100MHz)δ(ppm):192.5,150.7,146.4,138.3,134.4,129.8,129.3,128.3,127.5,126.4,124.1,116.1,38.9,23.7,23.1;IR(KBr,cm-1):ν3042,2955,1693,1665,1601,1500,1423,1167,1021,925,766;HRMS(ESI)m/z[M+H]+calcdforC17H14N2OS:295.0905;found:295.0903。(17)1-苯基-3-(3-噻唑基)-1,5,6,7-4H-四氢吲唑-4-酮(4q):分子结构式:实验数据:黄色固体;收率:63%;m.p.152.3-152.9°C(PE/EA);1HNMR(CDCl3,400MHz)δ(ppm):8.78(d,J=2.8Hz,1H),7.86(d,J=5.6Hz,1H),7.55-7.49(m,4H),7.43(dd,J=6.4,6.8Hz,1H),7.31(dd,J=4.8,2.8Hz,1H),2.95(t,J=6.0Hz,2H),2.60(t,J=6.0Hz,2H),2.17-2.11(m,2H);13CNMR(CDCl3,150MHz)δ(ppm):192.9,150.7,147.3,138.6,132.8,129.4,128.3,127.6,127.1,124.8,124.2,116.7,39.1,23.8,23.3;IR(KBr,cm-1):ν3043,2956,1695,1662,1600,1488,1429,1168,1023,927,761;HRMS(ESI)m/z[M+H]+calcdforC17H14N2OS:295.0905;found:295.0905。(18)1-苯基-3-(6-(2,3-二氢苯并[b][1,4]二噁烷基))-1,5,6,7-4H-四氢吲唑-4-酮(4r):分子结构式:实验数据:黄色固体;收率:45%;m.p.188.7-189.3°C(PE/EA);1HNMR(CDCl3,400MHz)δ(ppm):7.72(s,1H),7.66(d,J=8.4Hz,1H),7.55-7.48(m,4H),7.41(dd,J=6.8,6.8Hz,1H),6.91(d,J=8.4Hz,1H),4.26(s,4H),2.96(t,J=6.0Hz,2H),2.58(t,J=6.0Hz,2H),2.17-2.11(m,2H);13CNMR(CDCl3,150MHz)δ(ppm):192.8,151.4,150.8,144.4,143.1,138.6,129.3,128.2,125.3,124.1,122.5,118.0,116.8,116.6,64.5,64.2,39.0,23.8,23.3;IR(KBr,cm-1):ν3042,2961,1680,1601,1503,1451,1178,1024,936,882,763;HRMS(ESI)m/z[M+H]+calcdforC21H18N2O3:347.1396;found:347.1389。三、化合物4b和4p的单晶表征:1、1-苯基-3-对甲苯基-1,5,6,7-4H-四氢吲唑-4-酮(4b)和1-苯基-3-(2-噻唑基)-1,5,6,7-4H-四氢吲唑-4-酮(4p)的分子结构图见图1、2。2、1-苯基-3-对甲苯基-1,5,6,7-4H-四氢吲唑-4-酮(4b)和1-苯基-3-(2-噻唑基)-1,5,6,7-4H-四氢吲唑-4-酮(4p)的单晶数据见表2。表2:化合物4b和4p的晶体参数表由4b和4p的分子结构图、单晶数据表可见:化合物4b和4p单晶的培养证明了目标化合物在所构建的吲唑环上具有1,3-二芳基和4-羰基结构单元,进一步证明可以通过取代β-硝基芳基乙烯、取代1,3-环己二酮和取代苯肼为基本底物,在催化剂三乙烯二胺(DABCO)的作用下进行多组分一锅煮反应,能够有效地合成具有高度区域选择性的1,3-二芳基-1,5,6,7-四氢吲唑衍生物。本方法通过取代β-硝基芳基乙烯、取代1,3-环己二酮和取代苯肼在三乙烯二胺(DABCO)的作用下进行多组分一锅煮反应合成目标化合物,具有反应步骤简洁,后处理方便,产物区域选择性高,能够有效地构建吲唑类药物基本骨架。四、取代1,3-环己二酮取代、β-硝基芳基乙烯和取代苯肼的投料优化试验:反应式如下:表3:取代1,3-环己二酮、取代β-硝基芳基乙烯和取代苯肼的投料摩尔比优化试验对比表以上试验中,各例采用的催化剂三乙烯二胺和β-硝基芳基乙烯的投料摩尔比为0.75∶1经试验表明取代1,3-环己二酮取代、β-硝基芳基乙烯和取代苯肼的投料摩尔比从1∶1∶1到1.2∶1∶1.05都能够以相应收率获得目标化合物。其中取代1,3-环己二酮、取代β-硝基芳基乙烯和取代苯肼的最佳投料摩尔比为1.1∶1∶1.05。五、催化剂三乙烯二胺(DABCO)的用量优化试验:反应式如下:表4:β-硝基芳基乙烯和三乙烯二胺(DABCO)的投料摩尔比试验结果对比表以上各例中取代1,3-环己二酮、取代β-硝基芳基乙烯和取代苯肼的投料摩尔比分别为1.1∶1∶1.05。表4的结果表明:β-硝基芳基乙烯和三乙烯二胺(DABCO)的投料摩尔比对反应影响比较大,使用为0.25∶1摩尔比,目标化合物的收率为43%,很显然三乙烯二胺(DABCO)的使用量不能够促进反应充分进行,而0.5∶1摩尔比能够有效地促进反应,而三乙烯二胺(DABCO)和β-硝基芳基乙烯的投料摩尔比为0.75∶1已达到最佳。六、反应温度对合成收率的优化试验:反应式如下:表5:不同温度条件下反应的对比表由上表可见:常温反应不利于目标化合物形成。为提高产率,通过改变反应温度研究对反应的影响,发现升高温度至50℃、60℃产率较大,可达83%,继续提高反应温度产率反而开始下降,由此确定了此条件下最佳反应温度为50~60℃。七、溶剂对合成收率的优化试验:反应式如下:表6:不同溶剂的对比试验对比表表6结果表明:不同反应溶剂对反应影响较大,乙腈和甲醇有利于目标化合物的形成,而极性比较大的N,N-二甲基甲酰胺不利于目标化合物的形成。而二氯甲烷和甲苯作为在反应温度为50℃的条件下,反应难以有效发生。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1