带有主客体识别基团的有机三重态光敏剂及其制备方法与流程
本发明涉及光敏剂技术领域,具体涉及一种带有主客体识别基团的有机三重态光敏剂及其制备方法。
背景技术:
随着社会的快速发展,世界范围的能源匮乏时代已经悄然来临,化石燃料的大量使用导致的环境问题驱使人类越来越关注太阳能这种取之不尽用之不竭的安全环保型能源。提高太阳能的利用效率是各学科科学家的不懈追求。
上转换技术是能将低能量光子转变为高能量光子的一项技术,在提高太阳能利用效率领域具有重要价值,目前已在光伏、光催化、荧光生物成像等领域有着广泛的应用。实现上转换的方法很多,如使用稀土金属、双光子吸收染料及无机晶体等均可实现上转换。
尽管这些上转换方法有着各自的特点和特殊的应用领域,但也存在缺陷,如稀土材料的吸光能力较弱,工作波长不易调节,上转换量子效率低等;双光子荧光染料(TPA)需要高强度的激发光,一般需要MW cm-2级别,是太阳光强度的1000万倍。这些缺点使得这些技术在太阳能电池等新兴领域的应用受到限制。
三重态-三重态湮灭上转换(Triplet-triplet annihilation upconversion,简称TTA上转换),因其具有所需激发光能量密度小(太阳光可作为激发光源)、光敏剂对可见光的吸收能力强、上转换的工作波长能方便可调等优点而受到广泛关注。三重激发态,TTA:三重态-三重态湮灭(受体分子之间),1A*:受体的单重激发态。TTA上转换体系包含两部分:三重态光敏剂(能量给体)与三重态受体(湮灭剂),三重态光敏剂吸收激发光后到达其激发单重态,经过系间窜越(ISC),到达其激发三重态;处于激发三重态的光敏剂分子在光敏剂和受体分子接近时,通过碰撞将能量传递给受体分子的三重态,即发生三重态-三重态能量转移(TTET),从而使受体分子的三重激发态得到布居;两个处于三重激发态的受体分子碰撞,发生湮灭,以一定几率生成一个受体分子的单重激发态,另一个则回到基态;处于单重激发态(S1)的受体分子以辐射跃迁的形式回到基态(S0),发出上转换的荧光。
三重态光敏剂主要被用来作为能量给体引发一个新的光化学和光物理过程,如发生三重态-三重态能量传递或者电子传递等。理想的三重态光敏剂应该具有强的可见光吸收能力、高的ISC效率保证分子有较高的三重态量子产率,长的三重激发态寿命以保证有足够的时间发生能量传递、参与各种光物理、光化学反应等特点。分子三重激发态得到有效布局的三重态光敏剂在电致发光、磷光生物成像或者分子传感、光动力学治疗(PDT,敏化产生单线态氧,1O2)、光催化有机反应、三重态-三重态湮灭上转换等领域有着广泛用途。
从以上三重态湮灭上转换机理的介绍中可以看出,三重态光敏剂是该类上转换技术不可或缺的一部分,三重态光敏剂的光物理性质对三重态湮灭上转换效率起着重要作用。到目前为止,具有超分子结合位点的三重态光敏剂还未见报道。具有超分子结合位点的三重态光敏剂对于提高三重态湮灭上转换效率具有非常重要的影响。
从三重态湮灭上转换机理得知,发生上转换需要两个组成部分—三重态光敏剂与受体协同完成,三重态光敏剂受光激发后需扩散至受体附近方能传递能量,这在某些分子扩散受限的体系如固体材料中是非常致命的缺陷。
技术实现要素:
本发明的目的在于提供一种带有主客体识别基团的有机三重态光敏剂及其制备方法,以解决现有光敏剂转换效率低的问题。
本发明解决上述技术问题的技术方案如下:
一种带有主客体识别基团的有机三重态光敏剂,其具有如下结构:
其分子式为:C102H57BF2N8O3S。
一种带有主客体识别基团的有机三重态光敏剂的制备方法,包括:
(1)制备化合物F:
(11)利用化合物A和1,2-二溴乙烷合成化合物B,其中,化合物A为对羟基苯甲醛,化合物B的结构为:
(12)将化合物B于惰性气氛中与2,4-二甲基吡咯和三氟乙酸在遮光环境中反应,反应结束后,依次加入DDQ、三乙胺和BF3·OEt2继续反应,制得化合物C,化合物C的结构为:
(13)利用化合物C和N-碘代琥珀酰亚胺合成化合物D,化合物D的结构为:
(14)将化合物D与5-醛基-2-噻吩硼酸混合,然后加入K3PO4·3H2O,然后于惰性气氛中在催化剂Pd(OAc)2的作用下合成化合物E,化合物E的结构为:
(15)利用化合物E和NaN3合成化合物F,化合物F的结构为:
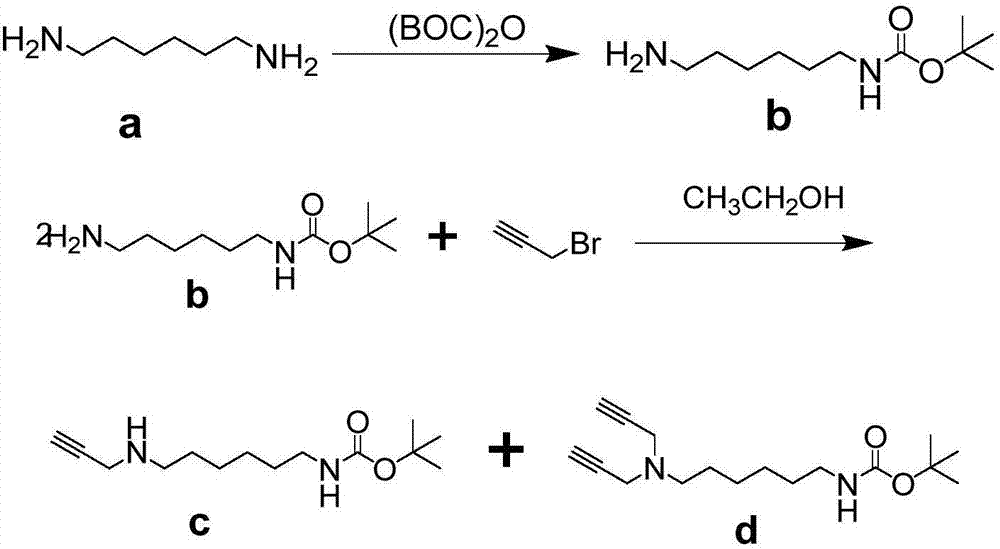
(2)制备化合物c:
(21)利用1,6-己二胺和(BOC)2O合成化合物b,化合物b的结构为:
(22)利用化合物b和3-溴丙炔制得化合物c,化合物c的结构为:
(3)将步骤(1)制得的化合物F与步骤(2)制得的化合物c混合,在惰性气氛中加入CuSO4·5H2O和抗坏血酸反应,制得化合物G,化合物G的结构为:
(4)将化合物G与C60和肌氨酸混合于惰性气氛中反应,制得有机三重态光敏剂。
进一步地,在本发明较佳的实施例中,制备化合物F包括以下具体过程:
(11)将羟基苯甲醛溶于乙醇中,然后加入1,2-二溴乙烷和碳酸钾在75-80℃的反应7.5-8.5h,利用薄层色谱法确认反应完全后,过滤,提纯,制得化合物B;其中,羟基苯甲醛、1,2-二溴乙烷和碳酸钾的摩尔比为1:(4.8-5.5):(1.0-1.2),薄层色谱法中采用的展开剂为二氯甲烷;
(12)在惰性气氛中,向反应器中加入化合物B和二氯甲烷,同时采用抽真空置换氮气的方式去除溶解在二氯甲烷中的氧气,然后加入2,4-二甲基吡咯和三氟乙酸在遮光环境中反应8-12h;利用薄层色谱法确认反应完全后,加入DDQ搅拌,反应0.8-1.5h后冷却反应体系;继续搅拌10-20min后加入三乙胺,继续搅拌0.4-0.6h后滴加BF3·OEt2,待反应完全后提纯,制得化合物C;其中,化合物B、2,4-二甲基吡咯和DDQ的摩尔比为1:2:1,三乙胺、BF3·OEt2与化合物B的摩尔比为10:10:1,三氟乙酸为催化量,薄层色谱法中采用的展开剂为二氯甲烷和正己烷按照1:1的体积比混合而成;
(13)将化合物C溶于二氯甲烷中,在冰水浴条件下,滴加溶有N-碘代琥珀酰亚胺的干燥二氯甲烷溶液,反应0.8-1.2h,用薄层色谱法确认反应完全后,提纯,制得化合物D;其中,化合物C与N-碘代琥珀酰亚胺的摩尔比为1:1,薄层色谱法中采用的展开剂为二氯甲烷和正己烷按照1:1的体积比混合而成;
(14)将化合物D和5-醛基-2-噻吩硼酸溶于甲苯和乙醇构成的混合溶剂中,搅拌均匀后加入K3PO4·3H2O,于惰性气氛中加入催化剂Pd(OAc)2在75-85℃下回流反应7.5-8.5h,用薄层色谱法确认反应完全后,停止加热,冷却至室温,提纯后制得化合物E;其中,化合物D、5-醛基-2-噻吩硼酸、K3PO4·3H2O与Pd(OAc)2的摩尔比为1:3:2:0.05,薄层色谱法中采用的展开剂为二氯甲烷和正己烷按照1:1的体积比混合而成;
(15)将NaN3溶于DMSO中,然后滴加化合物E反应8-12min,待反应完全后,提纯,制得化合物F;其中,化合物F与NaN3的摩尔比为0.4:1。
需要说明的是,在步骤(11)中,碳酸钾用于中和反应产物溴化氢;在步骤(12)中,二氯甲烷作为溶剂,其加量没有特殊限制,“遮光环境”也可以被理解为是黑暗环境或者暗箱。
进一步地,在本发明较佳的实施例中,在步骤(12)中,提纯包括以下具体过程:
用旋转蒸发仪去除反应产物中的溶剂,再用硅胶色谱柱进一步提纯,将提纯得到的一次粗产品用饱和碳酸氢钠溶液清洗,然后用二氯甲烷萃取,合并有机层,用硫酸钠干燥后,过滤,浓缩,将得到的二次粗产品再次用硅胶柱色谱提纯,得到化合物C;其中,硅胶柱采用的洗脱剂由二氯甲烷:石油醚按照1:1的体积比混合而成。
进一步地,在本发明较佳的实施例中,制备化合物c包括以下具体过程:
(21)将(BOC)2O溶于二氯甲烷中混合,2-3h后,将混合溶液滴加至冰水浴的1,6-己二胺中,于室温下反应23-25h,用展开剂确认反应完全后,提纯,制得化合物b;其中,1,6-己二胺与(BOC)2O的摩尔比为(6-8):1展开剂为二氯甲烷、甲醇和三乙胺按照100:10:1的体积比混合而成;
(22)将化合物b和3-溴丙炔溶于乙醇中,然后加入K2CO3,在搅拌条件下加热至35-45℃反应2.5-3.5h,用展开剂确认反应完全后,提纯,制得化合物c;其中,化合物b、3-溴丙炔以及K2CO3的摩尔比为1:1:(2.5-3),展开剂为二氯甲烷和甲醇按照20:1的体积比混合而成。
进一步地,在本发明较佳的实施例中,步骤(3)包括以下具体过程:
将化合物F和化合物c溶于二氯甲烷中,在惰性气氛下加入CuSO4·5H2O和抗坏血酸反应,用薄层色谱法确认反应完全后,取出产物并冷藏,制得化合物G;其中,化合物F、化合物c、CuSO4·5H2O和抗坏血酸的摩尔比为1:1:0.3:0.6,薄层色谱法中采用的展开剂为二氯甲烷和甲醇按照30:1的体积比混合而成。
进一步地,在本发明较佳的实施例中,步骤(4)包括以下具体过程:
将化合物G、C60和肌氨酸混合后,加入甲苯混合,将混合溶液在惰性气氛中升温至105-120℃回流反应1.5-2.5h,然后冷却至室温,提纯,制得有机三重态光敏剂;其中,化合物G、C60和肌氨酸的摩尔比为1:1:4。
在步骤(4)中,甲苯作为溶剂,其加量没有特别限制。本发明使用的C60的甲苯溶液,其浓度优选为1mg/ml。
进一步地,在本发明较佳的实施例中,惰性气氛通过抽真空置换氮气的方法获得。
本发明具有以下有益效果:
本发明的有机三重态光敏剂,其化学结构上带有主客体识别位点,通过该识别位点与结合有超分子的三重态受体发生主客体作用,能在扩散受限体系中拉近光敏剂与受体的空间距离,从而实现在扩散受限体系中提高上转换效率。
本发明合成的有机三重态光敏剂为未报道的新化合物,其具有主客体识别基团:Bodipy的中位苯环位置通过Click反应引入一端被叔丁氧羰基保护己二胺作为超分子结合位点,叔丁氧羰基将一端氨基保护的目的是为了提高光敏剂的稳定性,便于其保存,该保护基团在酸性条件下即可去除,酸性条件同时可使氨基质子化,而质子化后的己二胺与葫芦[6]脲具有强的主客体相互作用,包结常数>108M。该类光敏剂与修饰了上述主体分子的三重态受体结合时,便可提高三重态光敏剂与三重态受体之间的能量传递效率,进而提高三重态三重态湮灭上转换的效率。
本发明的有机三重态光敏剂通过在C60-Bodipy二元化合物有机三重态光敏剂中Bodipy的中位苯环位置上引入己二胺作为超分子结合位点,该位置对三重态光敏剂的光物理性质没有影响,而通过具有超子结合位点的三重态光敏剂与含有三重态受体的超分子主体在溶液中进行自组装,拉近三重态光敏剂与三重态受体之间的空间距离至分子尺寸范围内,使得具有较大分子尺寸的含C60的有机三重态光敏剂无须在有效寿命范围内扩散便能传递能量,从而降低三重态激子通过非辐射途径失活的概率,提高三重态光敏剂与三重态受体之间的能量传递效率,进而提高上转换效率。
本发明在合成关键中间体化合物C时,通过营造避光的反应环境,避免了该步骤中所用的原料之一:2,4-二甲基吡咯受光或者氧气而容易变质的情形、而导致产率降低;而且本发明严格控制化合物B和2,4-二甲基吡咯的投料比为1:2;在该两个条件的严格把控下,使得化合物C的产率明显提升。此外,对于后续的提纯步骤中,本发明用饱和碳酸氢钠水洗,从而将反应后剩余的用色谱柱很难分离掉的三氟化硼乙醚通过酸碱中和而除去,提高了化合物C的产品纯度,从而提高化合物C整体产品质量,为后续合成有机三重态光敏剂打下基础,为最终产物质量提供了保证。
附图说明
图1为化合物F的合成路线图;
图2为化合物c的合成路线图;
图3为有机三重态光敏剂:化合物H的合成路线图;
图4为化合物C的核磁共振光谱图;
图5为化合物D的核磁共振光谱图;
图6为化合物E的核磁共振光谱图;
图7为化合物F的核磁共振光谱图;
图8为化合物b的核磁共振光谱图;
图9为化合物c的核磁共振光谱图;
图10为化合物d的核磁共振光谱图;
图11为有机三重态光敏剂:化合物H的核磁共振光谱图。
具体实施方式
以下结合附图对本发明的原理和特征进行描述,所举实例只用于解释本发明,并非用于限定本发明的范围。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。
实施例1
本实施例的带有主客体识别基团的有机三重态光敏剂的制备方法,包括:
(1)制备化合物F:
合成路线如图1所示。
(11)利用化合物A和1,2-二溴乙烷合成化合物B。具体过程如下:
将羟基苯甲醛溶于乙醇中,然后加入1,2-二溴乙烷和碳酸钾在78℃的反应8h,利用薄层色谱法确认反应完全后,过滤,提纯,制得化合物B;其中,羟基苯甲醛、1,2-二溴乙烷和碳酸钾的摩尔比为1:5:1,薄层色谱法中采用的展开剂为二氯甲烷。本步骤提纯的具体过程为:用二氯甲烷萃取反应产物,合并有机相,用无水Na2SO4干燥并用旋转蒸发仪除去有机溶剂,将得到的粗产品用硅胶柱色谱提纯(洗脱剂:二氯甲烷)。
化合物B的产率为30.4%。
(12)将化合物B于惰性气氛中与2,4-二甲基吡咯和三氟乙酸在遮光环境中反应,反应结束后,先后加入DDQ、三乙胺和BF3·OEt2继续反应,制得化合物C。具体过程如下:
在惰性气氛中,向反应器中加入化合物B和干燥二氯甲烷,同时采用抽真空置换氮气的方式去除溶解在二氯甲烷中的氧气,然后加入2,4-二甲基吡咯和三氟乙酸在遮光环境中反应10h;利用薄层色谱法确认反应完全后,加入DDQ搅拌,反应1h后冷却反应体系;继续搅拌15min后加入三乙胺,继续搅拌0.5h后滴加BF3·OEt2,待反应完全后提纯,当有明显的荧光产物点产生后,表明反应完全,制得化合物C;其中,化合物B、2,4-二甲基吡咯和DDQ的摩尔比为1:2:1,三乙胺、BF3·OEt2与化合物B的摩尔比为10:10:1,三氟乙酸为催化量,薄层色谱法中采用的展开剂为二氯甲烷和正己烷按照1:1的体积比混合而成。本步骤提纯包括以下具体过程:用旋转蒸发仪去除反应产物中的溶剂,再用硅胶色谱柱进一步提纯,将提纯得到的一次粗产品用饱和碳酸氢钠溶液清洗,然后用二氯甲烷萃取,合并有机层,用硫酸钠干燥后,过滤,浓缩,将得到的二次粗产品再次用硅胶柱色谱提纯,得到化合物C;其中,硅胶柱采用的洗脱剂为二氯甲烷:石油醚按照1:1的体积比混合而成的。
化合物C为橙黄色,产率为11.4%。
(13)利用化合物C和N-碘代琥珀酰亚胺合成化合物D。具体过程如下:
将化合物C溶于干燥二氯甲烷中,在冰水浴条件下,滴加溶有N-碘代琥珀酰亚胺的干燥二氯甲烷溶液,反应1h,用薄层色谱法确认反应完全后,提纯,制得化合物D;其中,化合物C与N-碘代琥珀酰亚胺的摩尔比为1:1,薄层色谱法中采用的展开剂为二氯甲烷和正己烷按照1:1的体积比混合而成。本步骤提纯包括以下具体过程:用旋转蒸发仪除去反应产物的有机溶剂,将得到的粗产品用硅胶柱色谱分离提纯。
化合物D为橙黄色,产率为55.9%。
(14)将化合物D与5-醛基-2-噻吩硼酸混合,然后加入K3PO4·3H2O,然后于惰性气氛中在催化剂Pd(OAc)2的作用下合成化合物E。具体过程如下:
将化合物D和5-醛基-2-噻吩硼酸溶于甲苯和乙醇构成的混合溶剂中,搅拌均匀后加入K3PO4·3H2O,于惰性气氛中加入催化剂Pd(OAc)2在80℃下回流反应8h,用薄层色谱法确认反应完全后,停止加热,冷却至室温,提纯后制得化合物E;其中,化合物D、5-醛基-2-噻吩硼酸、K3PO4·3H2O与Pd(OAc)2的摩尔比为1:3:2:0.05,薄层色谱法中采用的展开剂为二氯甲烷和正己烷按照1:1的体积比混合而成。本步骤提纯包括以下具体过程:将反应得到的粗产物用硅胶柱色谱提纯,洗脱剂为二氯甲烷:正己烷=1:1。
化合物E为红色粉末,产率为14.2%。
(15)利用化合物E和NaN3合成化合物F。具体过程如下:
将NaN3溶于DMSO中,然后滴加化合物E反应10min,待反应完全后,提纯,制得化合物F;其中,化合物F与NaN3的摩尔比为0.4:1。本步骤提纯包括以下具体过程:确定原料基本反应完后,立刻加水水洗,此时溶液出现乳浊状,然后加入适量的NaCl去乳化,然后用CH2Cl2萃取,收集有机层用Na2SO4干燥后,旋干,粗产品用硅胶柱色谱分离提纯。
化合物F为红色产物,产率为85.4%。
(2)制备化合物c:
合成路线如图2所示。
(21)利用1,6-己二胺和(BOC)2O合成化合物b。具体过程如下:
将(BOC)2O溶于二氯甲烷中混合,2-3h后,将混合溶液滴加至冰水浴的1,6-己二胺中,于室温下反应24h,用展开剂确认反应完全后,提纯,制得化合物b;其中,1,6-己二胺与(BOC)2O的摩尔比为7.7:1,展开剂为二氯甲烷、甲醇和三乙胺按照100:10:1的体积比混合而成。本步骤提纯包括以下具体过程:向产物中加入适量水洗去未反应的1,6-己二胺,过滤,滤液用CH2Cl2萃取,把有机层用Na2SO4干燥后,用旋转蒸发仪除去溶剂,粗用产品胶柱色谱提纯(洗脱剂为二氯甲烷:甲醇:三乙胺=100:10:1),用茚三酮显色。
化合物b为无色液体,产率为17.9%。
(22)利用化合物b和3-溴丙炔制得化合物c。具体过程如下:
将化合物b和3-溴丙炔溶于乙醇中,然后加入K2CO3,在搅拌条件下加热至40℃反应3h,用展开剂确认反应完全后,提纯,制得化合物c;其中,化合物b、3-溴丙炔以及K2CO3的摩尔比为1:1:2.8,展开剂为二氯甲烷和甲醇按照20:1的体积比混合而成。本步骤提纯包括以下具体过程:用旋转蒸发仪除去产物中的有机溶剂,将得到的粗产品用硅胶柱色谱分离提纯(洗脱机为CH2Cl2:CH3OH=20:1)。
化合物c为产率26.6%。
(3)制备化合物G:
合成路线如图3所示。
将化合物F和化合物c溶于二氯甲烷中,在惰性气氛下加入CuSO4·5H2O和抗坏血酸反应,用薄层色谱法确认反应完全后,取出产物并冷藏,制得化合物G;其中,化合物F、化合物c、CuSO4·5H2O和抗坏血酸的摩尔比为1:1:0.3:0.6,,薄层色谱法中采用的展开剂为二氯甲烷甲醇按照30:1的体积比混合而成。
(4)化合物G与C60和肌氨酸反应
将化合物G、C60和肌氨酸混合后,加入甲苯混合,将混合溶液在惰性气氛中升温至110℃回流反应2h,然后冷却至室温,提纯,制得有机三重态光敏剂;其中,化合物G、C60和肌氨酸的摩尔比为1:1:4。本步骤提纯包括以下具体过程:用旋转蒸发仪出去产物溶剂,粗产品用硅胶柱色谱提纯,洗脱剂为甲苯:正己烷=5:1。
红色产物H为有机三重态光敏剂。
实施例2
本实施例与实施例1的区别在于各个步骤的反应条件略有不同,具体如下:
本实施例的带有主客体识别基团的有机三重态光敏剂的制备方法,包括:
(1)制备化合物F:
(11)利用化合物A和1,2-二溴乙烷合成化合物B。具体过程如下:
将羟基苯甲醛溶于乙醇中,然后加入1,2-二溴乙烷和碳酸钾在75℃的反应7.5h,利用薄层色谱法确认反应完全后,过滤,提纯,制得化合物B;其中,羟基苯甲醛、1,2-二溴乙烷和碳酸钾的摩尔比为1:4.8:1,薄层色谱法中采用的展开剂为二氯甲烷。
(12)将化合物B于惰性气氛中与2,4-二甲基吡咯和三氟乙酸在遮光环境中反应,反应结束后,先后加入DDQ、三乙胺和BF3·OEt2继续反应,制得化合物C。具体过程如下:
在惰性气氛中,向反应器中加入化合物B和干燥二氯甲烷,同时采用抽真空置换氮气的方式去除溶解在二氯甲烷中的氧气,然后加入2,4-二甲基吡咯和三氟乙酸在遮光环境中反应8h;利用薄层色谱法确认反应完全后,加入DDQ搅拌,反应0.8h后冷却反应体系;继续搅拌10min后加入三乙胺,继续搅拌0.4h后滴加BF3·OEt2,待反应完全后提纯,当有明显的荧光产物点产生后,表明反应完全,制得化合物C;其中,化合物B、2,4-二甲基吡咯和DDQ的摩尔比为1:2:1,三乙胺、BF3·OEt2与化合物B的摩尔比为10:10:1,三氟乙酸为催化量,薄层色谱法中采用的展开剂为二氯甲烷和正己烷按照1:1的体积比混合而成。
(13)利用化合物C和N-碘代琥珀酰亚胺合成化合物D。具体过程如下:
将化合物C溶于干燥二氯甲烷中,在冰水浴条件下,滴加溶有N-碘代琥珀酰亚胺的干燥二氯甲烷溶液,反应0.8h,用薄层色谱法确认反应完全后,提纯,制得化合物D;其中,化合物C与N-碘代琥珀酰亚胺的摩尔比为1:1,薄层色谱法中采用的展开剂为二氯甲烷和正己烷按照1:1的体积比混合而成。
(14)将化合物D与5-醛基-2-噻吩硼酸混合,然后加入K3PO4·3H2O,然后于惰性气氛中在催化剂Pd(OAc)2的作用下合成化合物E。具体过程如下:
将化合物D和5-醛基-2-噻吩硼酸溶于甲苯和乙醇构成的混合溶剂中,搅拌均匀后加入K3PO4·3H2O,于惰性气氛中加入催化剂Pd(OAc)2在75℃下回流反应7.5h,用薄层色谱法确认反应完全后,停止加热,冷却至室温,提纯后制得化合物E;其中,化合物D、5-醛基-2-噻吩硼酸、K3PO4·3H2O与Pd(OAc)2的摩尔比为1:3:2:0.05,薄层色谱法中采用的展开剂为二氯甲烷和正己烷按照1:1的体积比混合而成。
(15)利用化合物E和NaN3合成化合物F。具体过程如下:
将NaN3溶于DMSO中,然后滴加化合物E反应8min,待反应完全后,提纯,制得化合物F;其中,化合物F与NaN3的摩尔比为0.4:1。
(2)制备化合物c:
(21)利用1,6-己二胺和(BOC)2O合成化合物b。具体过程如下:
将(BOC)2O溶于二氯甲烷中混合,2h后,将混合溶液滴加至冰水浴的1,6-己二胺中,于室温下反应23h,用展开剂确认反应完全后,提纯,制得化合物b;其中,1,6-己二胺与(BOC)2O的摩尔比为6:1展开剂为二氯甲烷、甲醇和三乙胺按照100:10:1的体积比混合而成。
(22)利用化合物b和3-溴丙炔制得化合物c。具体过程如下:
将化合物b和3-溴丙炔溶于乙醇中,然后加入K2CO3,在搅拌条件下加热至35℃反应2.5h,用展开剂确认反应完全后,提纯,制得化合物c;其中,化合物b、3-溴丙炔以及K2CO3的摩尔比为1:1:2.5,展开剂为二氯甲烷和甲醇按照20:1的体积比混合而成。
(3)制备化合物G:
将化合物F和化合物c溶于二氯甲烷中,在惰性气氛下加入CuSO4·5H2O和抗坏血酸反应,用薄层色谱法确认反应完全后,取出产物并冷藏,制得化合物G;其中,化合物F、化合物c、CuSO4·5H2O和抗坏血酸的摩尔比为1:1:0.3:0.6,薄层色谱法中采用的展开剂为二氯甲烷甲醇按照30:1的体积比混合而成。
(4)化合物G与C60和肌氨酸反应
将化合物G、C60和肌氨酸混合后,加入甲苯混合,将混合溶液在惰性气氛中升温至105℃回流反应1.5h,然后冷却至室温,提纯,制得有机三重态光敏剂;其中,化合物G、C60和肌氨酸的摩尔比为1:1:4。
实施例3
本实施例与实施例1的区别在于各个步骤的反应条件略有不同,具体如下:
本实施例的带有主客体识别基团的有机三重态光敏剂的制备方法,包括:
(1)制备化合物F:
(11)利用化合物A和1,2-二溴乙烷合成化合物B。具体过程如下:
将羟基苯甲醛溶于乙醇中,然后加入1,2-二溴乙烷和碳酸钾在80℃的反应8.5h,利用薄层色谱法二氯甲烷确认反应完全后,过滤,提纯,制得化合物B;其中,羟基苯甲醛、1,2-二溴乙烷和碳酸钾的摩尔比为1:5.5:1.2,薄层色谱法中采用的展开剂为二氯甲烷。
(12)将化合物B于惰性气氛中与2,4-二甲基吡咯和三氟乙酸在遮光环境中反应,反应结束后,先后加入DDQ、三乙胺和BF3·OEt2继续反应,制得化合物C。具体过程如下:
在惰性气氛中,向反应器中加入化合物B和干燥二氯甲烷,同时采用抽真空置换氮气的方式去除溶解在二氯甲烷中的氧气,然后加入2,4-二甲基吡咯和三氟乙酸在遮光环境中反应12h;利用薄层色谱法确认反应完全后,加入DDQ搅拌,反应1.5h后冷却反应体系;继续搅拌20min后加入三乙胺,继续搅拌0.6h后滴加BF3·OEt2,待反应完全后提纯,当有明显的荧光产物点产生后,表明反应完全,制得化合物C;其中,化合物B、2,4-二甲基吡咯和DDQ的摩尔比为1:2:1,三乙胺、BF3·OEt2与化合物B的摩尔比为10:10:1,三氟乙酸为催化量,薄层色谱法中采用的展开剂为二氯甲烷和正己烷按照1:1的体积比混合而成。
(13)利用化合物C和N-碘代琥珀酰亚胺合成化合物D。具体过程如下:
将化合物C溶于干燥二氯甲烷中,在冰水浴条件下,滴加溶有N-碘代琥珀酰亚胺的干燥二氯甲烷溶液,反应1.2h,用薄层色谱法确认反应完全后,提纯,制得化合物D;其中,化合物C与N-碘代琥珀酰亚胺的摩尔比为1:1,薄层色谱法中采用的展开剂为二氯甲烷和正己烷按照1:1的体积比混合而成。
(14)将化合物D与5-醛基-2-噻吩硼酸混合,然后加入K3PO4·3H2O,然后于惰性气氛中在催化剂Pd(OAc)2的作用下合成化合物E。具体过程如下:
将化合物D和5-醛基-2-噻吩硼酸溶于甲苯和乙醇构成的混合溶剂中,搅拌均匀后加入K3PO4·3H2O,于惰性气氛中加入催化剂Pd(OAc)2在85℃下回流反应8.5h,用薄层色谱法确认反应完全后,停止加热,冷却至室温,提纯后制得化合物E;其中,化合物D、5-醛基-2-噻吩硼酸、K3PO4·3H2O与Pd(OAc)2的摩尔比为1:3:2:0.05,薄层色谱法中采用的展开剂为二氯甲烷和正己烷按照1:1的体积比混合而成。
(15)利用化合物E和NaN3合成化合物F。具体过程如下:
将NaN3溶于DMSO中,然后滴加化合物E反应12min,待反应完全后,提纯,制得化合物F;其中,化合物F与NaN3的摩尔比为0.4:1。
(2)制备化合物c:
(21)利用1,6-己二胺和(BOC)2O合成化合物b。具体过程如下:
将(BOC)2O溶于二氯甲烷中混合,3h后,将混合溶液滴加至冰水浴的1,6-己二胺中,于室温下反应25h,用展开剂确认反应完全后,提纯,制得化合物b;其中,1,6-己二胺与(BOC)2O的摩尔比为8:1展开剂为二氯甲烷、甲醇和三乙胺按照100:10:1的体积比混合而成。
(22)利用化合物b和3-溴丙炔制得化合物c。具体过程如下:
将化合物b和3-溴丙炔溶于乙醇中,然后加入K2CO3,在搅拌条件下加热至45℃反应3.5h,用展开剂确认反应完全后,提纯,制得化合物c;其中,化合物b、3-溴丙炔以及K2CO3的摩尔比为1:1:3,展开剂为二氯甲烷和甲醇按照20:1的体积比混合而成。
(3)制备化合物G:
将化合物F和化合物c溶于二氯甲烷中,在惰性气氛下加入CuSO4·5H2O和抗坏血酸反应,用薄层色谱法确认反应完全后,取出产物并冷藏,制得化合物G;其中,化合物F、化合物c、CuSO4·5H2O和抗坏血酸的摩尔比为1:1:0.3:0.6,薄层色谱法中采用的展开剂为二氯甲烷和甲醇按照30:1的体积比混合而成。
(4)化合物G与C60和肌氨酸反应
将化合物G、C60和肌氨酸混合后,加入甲苯混合,将混合溶液在惰性气氛中升温至120℃回流反应2.5h,然后冷却至室温,提纯,制得有机三重态光敏剂;其中,化合物G、C60和肌氨酸的摩尔比为1:1:4。
实施例4
本实施例的带有主客体识别基团的有机三重态光敏剂的制备方法,包括:
1.化合物B的合成
在三口烧瓶中将对羟基苯甲醛(6.0g,49.2mmol)溶解于60mL乙醇中,加入1,2-二溴乙烷(46.2g,21.0mL,246.0mmol)和碳酸钾(6.6g,47.0mmol)在充分搅拌后加热78℃反应,大约反应8小时。TCL(二氯甲烷为展开剂)确认对羟基苯甲醛基本反应完全后,过滤,然后用二氯甲烷萃取,合并有机相,用无水Na2SO4干燥并用旋转蒸发仪除去有机溶剂,得到的粗产品用硅胶柱色谱提纯(洗脱剂:二氯甲烷),得到产物B(3.4g)。产率:30.4%。
2.化合物C的合成
用抽真空置换氮气的方法除去反应体系中的氧气,在氮气保护下加入化合物B(2.3g,10.0mmol)到三口烧瓶中,再加入400.0mL用分子筛干燥的二氯甲烷,再抽真空置换氮气的方法除去二氯甲烷中溶解的氧气,充分除去氧气后用5mL注射器取2,4-二甲基吡咯(2.2mL,20.0mmol)注射入反应瓶中,然后再用同样的方法加入三氟乙酸0.2mL,在橡胶塞的针孔处涂上真空脂,然后用锡箔纸把反应体系包住遮光反应一个晚上。TCL(展开剂:二氯甲烷)确认原料反应完后加入DDQ(2.3g,10.0mmol),搅拌1小时,然后用冰水浴冷却反应体系,搅拌15分钟后下加入12mL三乙胺,继续搅拌0.5h,再用滴管逐滴加入12.0mL BF3·OEt2,继续反应一段时间,确认反应中间体原料差不多反应完并且有明显的荧光产物点后停止反应,然后用旋转蒸发仪除去一部分溶剂后用硅胶柱色谱粗提纯,除去大量的杂质,再把得到的粗产品用饱和NaHCO3溶液水洗后用二氯甲烷萃取,合并有机层,用Na2SO4干燥后,过滤,浓缩,粗产品用硅胶柱色谱提纯,洗脱剂为(二氯甲烷:石油醚=1:1),得到橙黄色产物C(510.0mg),产率为11.4%。
化合物C:1HNMR(400MHz,CDCl3):7.20(d,2H,J=8.64Hz),7.04(d,2H,J=8.62Hz),6.00(s,2H),4.38(t,2H,J=6.21Hz),3.71(t,2H,J=6.23Hz),2.57(s,6H),1.45(s,6H).
3.化合物D的合成
将化合物C(200.0mg,44.6mmol)溶解于30mL分子筛干燥的CH2Cl2中,冰水浴条件下,用恒压滴液漏斗把溶解有N-碘代琥珀酰亚胺(NIS)(100.4mg,44.6mmol)的50mL分子筛干燥的CH2Cl2溶液缓慢滴加入到化合物C溶液中,约1h滴加完成,继续反应一个小时,然后用TCL(展开剂为二氯甲烷:正己烷=1:1)确认原料反应完全后。用旋转蒸发仪除去有机溶剂,粗产品用硅胶柱色谱分离提纯,得到橙黄产物D(143.0mg),产率:55.9%。
化合物D:1HNMR(400MHz,CDCl3):7.19(d,2H,J=8.65Hz),7.05(d,2H,J=8.66Hz),6.07(s,1H),4.39(t,2H,J=6.22Hz),3.72(t,2H,J=6.25Hz),2.65(s,3H),2.59(s,H),1.46(s,6H).
4.化合物E的合成
将5-醛基-2-噻吩硼酸(228.7mg,1.5mmol)与中间体D(251.9mg,0.5mmol)溶解于10mL甲苯与10mL乙醇的混合溶剂中,充分搅拌后加入K3PO4·3H2O(260mg,1.0mmol),然后使用抽真空—置换氮气的除氧方法,将此操作循环3~5次充分出去系统中的氧气后加入催化剂Pd(OAc)2(6.4mg,0.03mmol),然后混合液加热至80℃回流反应8小时,TCL(展开剂为二氯甲烷:正己烷=1:1)确定原料基本反应完后,停止反应,冷却至室温,粗产物用硅胶柱色谱提纯,洗脱剂为二氯甲烷:正己烷=1:1,得到红色粉末产物(544.0mg),产率:14.2%。
化合物E:1HNMR(400MHz,CDCl3):9.87(s,1H),7.73(d,1H,J=3.54),7.20(d,2H,J=8.05Hz),7.04(d,2H,J=8.02Hz),6.94(d,1H,J=3.51Hz),6.07(s,1H),4.36(t,2H,J=6.16Hz),3.69(t,2H,J=6.11Hz),2.59(d,6H,J=8.16Hz),1.46(s,6H).
5.化合物F的合成
将NaN3(15.2mg,0.2mmol)溶解于10mL DMSO中,然后滴加至中间体E(44.0mg,0.08mmol)中反应约10min左右,确定原料基本反应完后,立刻加水水洗,溶液出现乳浊状,加入适量的NaCl去乳化,然后用CH2Cl2萃取,收集有机层用Na2SO4干燥后,旋干,粗产品用硅胶柱色谱分离提纯。最后得到红色产物F(34.6mg),产率:85.4%。
化合物F:1HNMR(400MHz,CDCl3):9.89(s,1H),7.75(d,1H,J=3.85Hz),7.23(d,2H,J=8.61Hz),7.08(d,2H,J=8.62Hz),6.97(d,1H,J=3.83Hz),6.10(s,1H),4.24(t,2H,J=5.03Hz),3.69(t,2H,J=4.91Hz),2.62(d,6H,J=8.28Hz),1.48(d,6H,J=2.02Hz).
6.化合物b的合成
将(BOC)2O(1.0g,4.6mmol)溶解于12mL CH2Cl2中约在2.5h滴加入冰水浴的1,6-己二胺(4.1g,35.6mmol)中,然后在室温下反应24h,TCL(展开剂为二氯甲烷:甲醇:三乙胺=100:10:1)确定反应原料a反应到一定程度后,然后加入适量水洗去未反应的1,6-己二胺,过滤,滤液用CH2Cl2萃取,把有机层用Na2SO4干燥后,用旋转蒸发仪除去溶剂,粗用产品胶柱色谱提纯(洗脱剂为二氯甲烷:甲醇:三乙胺=100:10:1),用茚三酮显色。分离提纯得到无色液体b(1.4g),产率为17.9%。
化合物b:1HNMR(400MHz,CDCl3):4.59(s,1H),3.10(d,2H,J=6.08Hz),2.70(t,2H,J=7.01Hz),2.11(s,2H),1.48-1.44(m,3H),1.39-1.28(m,4H).
7.化合物c、d的合成
把中间体b(1.3g,6.4mmol)与3-溴丙炔(0.8g,6.4mmol)按1:1的当量溶解在20mL的乙醇中,同时加入K2CO3(2.5g,18.2mmol)。在搅拌的条件下加热至40℃反应3小时,TCL(展开剂为二氯甲烷:甲醇=20:1)确认原料反应完全之后停止反应,用旋转蒸发仪除去有机溶剂,粗产品用硅胶柱色谱分离提纯(洗脱机为CH2Cl2:CH3OH=20:1),收集得到双边产物d(274.0mg),产率:33.1%;得到单边产物c(200.0mg),产率:26.6%.
8.化合物G的合成
将中间产物F(34.6mg,0.07mmol)与中间产物c(16.0mg,0.07mmol)按1:1的当量溶解在6mL的CH2Cl2中,然后抽真空-置换氮气除去氧气,在氮气保护下加入CuSO4·5H2O(5mg)和抗坏血酸(8mg)。展开剂为(CH2Cl2:MeOH=30:1),确定原料反应完后停止反应,在室温下旋干点板确定产物G较纯后溶解在CH2Cl2中放置冰箱保存待用(由于产物不太稳定,容易变质所以未能称出质量)
9.化合物H的合成
将上述中间产物G(0.05mmol),C60(37.7mmol,0.05mmol)和肌氨酸(16.2mg,0.2mmol)加入到100mL二口瓶中,在加入35mL甲苯,然后抽真空-置换氮气三次,在氮气保护下,混合液缓慢升温至110℃,回流反应2h,然后降至室温,用旋转蒸发仪出去溶剂,粗产品用硅胶柱色谱提纯,洗脱剂为甲苯:正己烷=5:1。得到红色产物H.
图4至图11为上述实施例4在各个步骤合成的化合物的核磁共振光谱图,根据图谱确定各化合物的化学结构。
本发明制备的化合物H拟用作三重态光敏剂,与连有超分子的三重态受体发生主客体相互作用,从而实现在扩散受限体系中实现三重态湮灭上转换。本发明的有机三重态光敏剂在应用时,将其用三氟乙酸在室温下进行酸化处理,然后再与连有超分子的三重态受体结合。酸化后过量的三氟乙酸通过旋转蒸发仪除去。
以上所述仅为本发明的较佳实施例,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种光敏剂制备复合光催化剂的方法及用途
- 一种可见光响应光敏剂负载纳米TiO<sub>2</sub>改性水性聚氨酯亮光漆的制备的制作方法
- 一种可见光响应光敏剂负载纳米TiO<sub>2</sub>改性水性聚氨酯亚光漆的制备的制作方法
- 凹凸棒石负载光敏剂的制备方法及紫外光固化涂料的制作方法
- 以硅胶为载体的固相光敏剂将速甾醇及其衍生物光敏异构化为预维生素d及其衍生物的方法
- 水溶性萘菁基化合物、制备方法及作为光敏剂的应用的制作方法
- 竹红菌乙素及其胺基衍生物类光敏剂的用途的制作方法
- 新型光敏剂和光伏器件的制作方法
- 用交联高分子光敏剂敏化制备cis-维生素D或其衍生物的方法
- 利用交联高分子光敏剂将速甾醇及其衍生物光敏异构化为预维生素d及其衍生物的方法