咪唑类衍生物及其制备方法与应用与流程
本发明涉及咪唑类衍生物及其制备方法与应用。
(二)背景技术
发光材料广泛应用于通讯、卫星、雷达、显示、生物分子探针等高科技领域。特别是在已进入信息时代的今天,满足各种信息显示需求的发光材料发展尤为迅速。近年来,具有π共轭结构的杂环化合物(如噻吩、噁二唑及咔唑衍生物等)作为发光材料,越来越引起人们的关注。其中氮杂环类化合物因其分子结构中的氮原子上的孤对电子参与了π共轭体系,通常表现出优异的发光性能。
咪唑类化合物,常存在于生物活性化合物以及天然产物中,一些咪唑类衍生物具备着抗菌、消炎、抗病毒、抗肿瘤的活性,在生物、医药领域有着重要的研究价值与应用前景。作为一种重要的五元氮杂环结构,其化学结构稳定,易于修饰。咪唑的π环中含有一个类吡啶氮原子和一个类吡咯氮原子,特殊的双“n”五元共轭体系赋予了该类化合物优异的光电性能。特别是苯并咪唑、菲并咪唑,其衍生物具有较高的发光效率、优异的光热稳定性、相对平衡的载流子注入\传输能力,在发光材料领域展现了较大的应用潜力。另外,以咪唑环为桥所构筑的共轭材料,在非线性光学、电化学、有机光电领域也显示出了很多独特的性能。
因此,设计合成新型咪唑环有机发光材料既能够丰富该结构的发光材料体系,又可以为充分研究咪唑类发光体系的光物理过程提供理论依据。
(三)
技术实现要素:
本发明设计合成了一种新型的咪唑类有机生色团,并对其基本光学性质进行了研究。
本发明的技术方案如下:
式(a)或式(b)所示的咪唑类衍生物:
式(a)或式(b)中,r=h、cn、f或ch3。
所述式(a)、式(b)所示的咪唑类衍生物的制备方法为:
(1)氮气保护下,将式(v)所示2-羟基-1,4-萘醌、苯胺、苯甲醛溶解在氯仿中,升温至回流状态(60℃),加入对甲基苯磺酸(催化剂),继续回流(60℃)反应6~8h,之后反应液经后处理,得到式(vi)所示化合物;
所述式(v)所示2-羟基-1,4-萘醌、苯胺、苯甲醛、对甲基苯磺酸的物质的量之比为1:1:1~5:0.2~0.25;
所述氯仿的体积用量以式(v)所示2-羟基-1,4-萘醌的质量计为2~5ml/g;
所述后处理的方法为:反应结束后,待反应液冷却至室温(20~30℃),加入水和二氯甲烷进行萃取,收集有机相,经无水硫酸镁干燥,减压浓缩,再进行柱层析纯化,以300~400目硅胶为固定相,二氯甲烷/石油醚体积比2:1的混合溶剂为流动相进行洗脱,收集含目标化合物的洗脱液,减压蒸除溶剂并干燥,得到式(vi)所示化合物;
(2)氮气保护下,将式(vi)所示化合物、式(vii)所示苯甲醛、醋酸铵和醋酸混合,升温至120~130℃反应12h,之后反应液经后处理,得到式(viii)所示化合物;
式(vii)或(viii)中,r的定义与上述相同;
所述式(vi)所示化合物、式(vii)所示苯甲醛、醋酸铵的物质的量之比为1:1~1.3:5;
所述醋酸的体积用量以式(vi)所示化合物的质量计为20~70ml/g;
所述后处理的方法为:反应结束后,待反应液冷却至室温,用饱和碳酸铵水溶液淬灭反应,再加入二氯甲烷萃取,收集有机相,经无水硫酸镁干燥,减压浓缩,再进行柱层析纯化,以300~400目硅胶为固定相,石油醚/二氯甲烷体积比1:4的混合溶剂为流动相进行洗脱,收集含目标化合物的洗脱液,减压蒸除溶剂并干燥,得到式(viii)所示化合物;
(3)氮气保护下,将式(viii)所示化合物、氢化钠、碘甲烷溶解于无水dmf(n,n-二甲基甲酰胺)中,室温反应12h,之后反应液经后处理,分离得到产物式(a)、式(b)所示的咪唑类衍生物;
所述式(viii)所示化合物、氢化钠、碘甲烷的物质的量之比为1:1.5:1.5;
所述dmf的体积用量以式(viii)所示化合物的质量计为20~50ml/g;
所述后处理的方法为:反应结束后,用饱和碳酸铵水溶液淬灭反应,再加入二氯甲烷萃取,收集有机相,经无水硫酸镁干燥,减压浓缩,再进行柱层析纯化,以300~400目硅胶为固定相,石油醚/二氯甲烷体积比1:4的混合溶剂为流动相进行洗脱,分别收集含目标化合物式(a)、式(b)的洗脱液,减压蒸除溶剂并干燥,得到式(a)、式(b)所示的咪唑类衍生物。
本发明通过核磁共振(nmr)、质谱(esi)表征了目标产物。
本发明所述式(a)或式(b)所示的咪唑类衍生物可作为光致发光材料应用。
本发明的有益效果在于:实现了通过非对称中间体合成稠环咪唑类化合物,且同时获得其两种同分异构产物。一个反应过程获得两种不同发光的分子,极大的丰富了基于咪唑环类结构的发光材料体系。
(四)附图说明
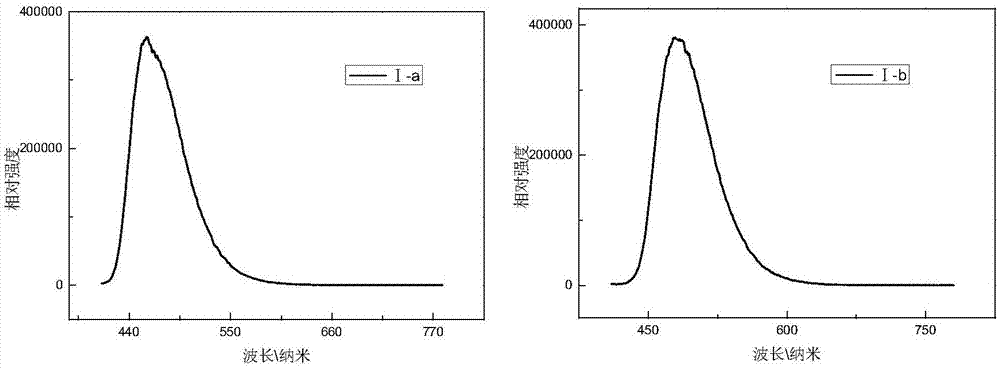
图1:本发明实施例5中化合物(i-a)和(i-b)的荧光发射曲线。
(五)具体实施方式
下面以具体实施例对本发明的技术方案作进一步说明,但本发明的保护范围并不仅限于此。
实施例13-甲基-2,13-二苯基-3h-苯并[c]咪唑并[4,5-a]吖啶(i-a),1-甲基-2,13-二苯基-1h-苯并[c]咪唑并[4,5-a]吖啶(i-b)的合成
(1)氮气保护下,将2-羟基-1,4-萘醌(3.00g,17mmol),苯甲醛(5.40g,51mmol),苯胺(1.6g,17mmol)溶解在氯仿(5ml)中,升温至60℃回流状态下加入催化剂对甲基苯磺酸(0.64g,3mmol),并于60℃下反应6小时。待体系冷却后用去离子水和二氯甲烷萃取,所得有机相加入无水mgso4干燥后,减压浓缩,再用柱层析分离提纯,固定相为300-400目硅胶,流动相为二氯甲烷/石油醚(体积比2:1),最后得到橙色固态的中间体7-苯基苯并[c]吖啶-5,6-二酮(vi)1.1g,产率为19%。1hnmr(cdcl3)9.04(d,1h,j=7.9hz),8.22-8.17(m,2h),7.90-7.83(m,2h),7.62(td,1h,j=7.8,0.9hz),7.57-7.53(m,3h),7.52-7.46(m,2h),7.26-7.23(m,2h);hrms(esi)[m+na]+:358.0844。
(2)氮气保护下,将化合物7-苯基苯并[c]吖啶-5,6-二酮(vi)1.1g(335.35g/mol,1.5mmol,0.50g)、苯甲醛(106.12g/mol,1.5mmol,0.159g)、醋酸铵(7.5mmol,0.578g)加入到100ml两口烧瓶中,加入醋酸35ml,温度升至120℃,反应12h。反应结束后,待体系冷却至室温,用饱和碳酸铵水溶液淬灭反应,再加入二氯甲烷萃取,合并有机相,以无水硫酸镁干燥过夜,抽滤除去干燥剂后,蒸除溶剂,重新加入二氯甲烷溶解,加入粗硅胶拌样,以层析法过柱,洗脱剂为石油醚与二氯甲烷混合溶液(体积比,1:4),最终得到黄色中间产物2,13-二苯基-1h-苯并[c]咪唑并[4,5-a]吖啶(viii)0.38g,产率为61%。确认物质的表征结构如下:1hnmr(500mhz,chloroform-d)δ9.60(d,j=8.1hz,1h),8.68(d,j=7.6hz,1h),8.45(d,j=8.3hz,1h),8.25(s,1h),7.91–7.79(m,5h),7.74(ddd,j=8.3,7.0,1.4hz,1h),7.69–7.59(m,5h),7.53(ddd,j=8.5,6.6,1.3hz,1h),7.47–7.37(m,3h);c30h19n3的ms(esi)数据表征实测为[m+h]+:422.1654。
(3)氮气保护下,将化合物2,13-二苯基-1h-苯并[c]咪唑并[4,5-a]吖啶(421.49g/mol,0.7mmol,0.30g)、氢化钠(1.06mmol,0.045g)、碘甲烷(1.06mmol,0.150g)加入到100ml两口烧瓶中,加入无水dmf15ml,室温反应12h。反应结束后,用饱和碳酸铵水溶液淬灭反应,再加入二氯甲烷萃取,合并有机相,以无水硫酸镁干燥过夜,抽滤除去干燥剂后,蒸除溶剂,重新加入二氯甲烷溶解,加入粗硅胶拌样,以层析法过柱,洗脱剂为石油醚与二氯甲烷混合溶液(体积比,1:4),最终得到一对同分异构体黄色目标产物3-甲基-2,13-二苯基-3h-苯并[c]咪唑并[4,5-a]吖啶(i-a)(0.23g),产率为74%。确认物质的表征结构如下:1hnmr(500mhz,dmso-d6)δ9.70(dd,j=8.2,1.2hz,1h),8.57(d,j=7.9hz,1h),8.38(d,j=8.5hz,1h),7.93-7.86(m,2h),7.81(td,j=7.7,7.2,1.0hz,1h),7.66–7.58(m,2h),7.61–7.52(m,5h),7.51–7.46(m,3h),7.42(dd,j=7.7,1.6hz,2h),4.29(s,3h);c31h21n3的ms(esi)数据表征实测为[m+h]+:436.1825。
1-甲基-2,13-二苯基-1h-苯并[c]咪唑并[4,5-a]吖啶(i-b)(0.060g),产率为19%。确认物质的表征结构如下1hnmr(500mhz,chloroform-d)δ9.53(d,j=8.0hz,1h),8.73(s,1h),8.44(d,j=8.4hz,1h),8.24–8.11(m,1h),7.84(dd,j=7.8,6.2hz,2h),7.71(ddt,j=20.9,16.0,7.0hz,8h),7.63–7.55(m,1h),7.54–7.45(m,3h),2.63(s,3h)。c31h21n3的ms(esi)数据表征实测为[m+h]+:436.18。
实施例24-(3-甲基-13-苯基-3h-苯并[c]咪唑并[4,5-a]吖啶-2-基)苄腈(ii-a),4-(1-甲基-13-苯基-1h-苯并[c]咪唑并[4,5-a]吖啶-2-基)苄腈(ii-b)的合成
合成方法同实施例1,不同之处在于,步骤(2)中,将苯甲醛替换为4-氰基苯甲醛(132g/mol,2mmol,0.264g),步骤(3)中将2,13-二苯基-1h-苯并[c]咪唑并[4,5-a]吖啶替换为4-(3-甲基-13-苯基-3h-苯并[c]咪唑并[4,5-a]吖啶-2-基)苄腈(446g/mol,0.67mmol,0.30g)。确认物质的表征结构如下1hnmr(500mhz,chloroform-d)δ9.61(d,j=7.9hz,1h),8.65(d,j=7.7hz,1h),8.48(d,j=8.5hz,1h),8.31(s,1h),7.92–7.81(m,5h),7.77(ddd,j=8.4,7.0,1.4hz,1h),7.73–7.64(m,7h),7.57(ddd,j=8.3,6.7,1.2hz,1h);c31h18n4的ms(esi)数据表征实测为[m+h]+:447.1604。
最终得到一对同分异构体黄色目标产物4-(3-甲基-13-苯基-3h-苯并[c]咪唑并[4,5-α]吖啶-2-基)苄腈(ii-a)(0.2g),产率为65%。确认物质的表征结构如下1hnmr(500mhz,chloroform-d)δ9.88(s,1h),8.46(s,1h),8.36(d,j=7.5hz,1h),7.87–7.76(m,4h),7.71(d,j=8.1hz,2h),7.63(d,j=8.1hz,2h),7.61–7.45(m,6h),4.32(s,3h);c32h20n4的ms(esi)数据表征实测为[m+h]+:461.1761。
4-(1-甲基-13-苯基-1h-苯并[c]咪唑并[4,5-a]吖啶-2-基)苄腈(ii-b)(0.050g),产率为16%。确认物质的表征结构如下:1hnmr(500mhz,chloroform-d)δ9.53(d,j=8.2hz,1h),8.67(d,j=8.3hz,1h),8.46(d,j=8.6hz,1h),8.16(d,j=8.5hz,1h),7.89–7.82(m,2h),7.81–7.70(m,10h),7.60(ddd,j=8.4,6.7,1.3hz,1h),2.65(s,3h);c32h20n4的ms(esi)数据表征实测为[m+h]+:461.1761。
实施例32-(4-氟苯基)-3-甲基-13-苯基-3h-苯并[c]咪唑并[4,5-a]吖啶(iii-a),2-(4-氟苯基)-1-甲基-13-苯基-1h-苯并[c]咪唑并[4,5-a]吖啶(iii-b)的合成
合成方法同实施例1,不同之处在于,步骤(2)中,将苯甲醛替换为4-氟苯甲醛(124.11g/mol,2mmol,0.250g),步骤(3)中将2,13-二苯基-1h-苯并[c]咪唑并[4,5-a]吖啶替换为2-(4-氟苯基)-13-苯基-1h-苯并[c]咪唑并[4,5-a]吖啶(439g/mol,0.68mmol,0.30g)。确认物质的表征结构如下:1hnmr(500mhz,chloroform-d)δ9.62(d,j=8.1hz,1h),8.67(d,j=7.7hz,1h),8.47(d,j=8.4hz,1h),8.20(s,1h),7.92–7.81(m,5h),7.76(ddd,j=8.3,7.1,1.4hz,1h),7.72–7.63(m,3h),7.65–7.57(m,2h),7.56(ddd,j=8.5,6.6,1.2hz,1h),7.14(t,2h);c30h18fn3的ms(esi)数据表征实测为[m+h]+:440.1558。
最终得到一对同分异构体黄色目标产物2-(4-氟苯基)-3-甲基-13-苯基-3h-苯并[c]咪唑并[4,5-a]吖啶(iii-a)(0.2g),产率为64%。确认物质的表征结构如下1hnmr(500mhz,chloroform-d)δ9.87(s,1h),8.43(s,1h),8.38(d,j=7.5hz,1h),7.80(m,4h),7.63–7.54(m,3h),7.54–7.47(m,5h),7.13(t,j=8.7hz,2h),4.30(s,3h);c31h20fn3的ms(esi)数据表征实测为[m+h]+:454.1714。
2-(4-氟苯基)-1-甲基-13-苯基-1h-苯并[c]咪唑并[4,5-a]吖啶(iii-b)(0.050g),产率为16%。确认物质的表征结构如下1hnmr(500mhz,chloroform-d)δ9.53(d,j=8.1hz,1h),8.68(s,1h),8.44(d,j=8.3hz,1h),8.15(d,j=8.7hz,1h),7.87–7.80(m,2h),7.76–7.68(m,6h),7.65(dd,j=8.5,5.4hz,2h),7.58(ddd,j=8.7,6.6,1.3hz,1h),7.19(t,j=8.6hz,2h),2.61(s,3h);c31h20fn3的ms(esi)数据表征实测为[m+h]+:454.1714。
实施例43-甲基-13-苯基-2-(对甲苯基-3h-苯并[c]咪唑并[4,5-a]吖啶(iv-a),1-甲基-13-苯基-2-(对甲苯基)-1h-苯并[c]咪唑并[4,5-a]吖啶(iv-b)的合成
合成方法同实施例1,不同之处在于,步骤(2)中,将苯甲醛替换为4-甲基苯甲醛(120.15g/mol,2mmol,0.245g),步骤(3)中将2,13-二苯基-1h-苯并[c]咪唑并[4,5-a]吖啶替换为13-苯基-2-(对甲苯基)-1h-苯并[c]咪唑并[4,5-a]吖啶(435g/mol,0.69mmol,0.30g)。确认物质的表征结构如下1hnmr(500mhz,chloroform-d)δ9.62(d,j=8.1hz,1h),8.70(d,j=7.7hz,1h),8.47(d,j=8.4hz,1h),8.24(s,1h),7.88–7.80(m,5h),7.76(ddd,j=8.3,7.0,1.4hz,1h),7.72–7.64(m,3h),7.58–7.54(m,1h),7.52(d,j=8.1hz,2h),7.26(d,j=7.6hz,2h),2.41(s,3h);c31h21n3的ms(esi)数据表征实测为[m+h]+:436.1808。
最终得到一对同分异构体黄色目标产物3-甲基-13-苯基-2-(对甲苯基)-3h-苯并[c]咪唑并[4,5-a]吖啶(iv-a)(0.2g),产率为65%,确认物质的表征结构如下1hnmr(500mhz,chloroform-d)δ9.86(d,j=7.7hz,1h),8.46(s,1h),8.37(d,j=7.7hz,1h),7.83–7.72(m,4h),7.63–7.53(m,3h),7.53–7.47(m,3h),7.42(d,j=8.1hz,2h),7.24(d,j=7.8hz,2h),4.29(s,3h),2.42(s,3h)。c32h23n3的ms(esi)数据表征实测为[m+h]+:450.1965。
1-甲基-13-苯基-2-(对甲苯基)-1h-苯并[c]咪唑并[4,5-a]吖啶(iv-b)(0.050g),产率为17%。确认物质的表征结构如下1hnmr(500mhz,chloroform-d)δ9.51(d,j=8.0hz,1h),8.68(d,j=7.7hz,1h),8.42(d,j=8.4hz,1h),8.14(d,j=8.6hz,1h),7.81(td,j=7.1,3.4hz,2h),7.74–7.64(m,6h),7.59–7.54(m,1h),7.52(d,j=7.9hz,2h),7.28(d,j=8.1hz,2h),2.59(s,3h),2.41(s,3h);c32h23n3的ms(esi)数据表征实测为[m+h]+:450.1965。
实施例5
取4.4mg(i-a)、(i-b)各自溶解于四氢呋喃中,并定容至10ml,配制成10-3mol/l的溶液用于测试。分别取30ul(i-a)、(i-b)溶液于比色皿,加入四氢呋喃2970ul稀释至10-5mol/l,依次测试荧光发射光谱。
该实施例可以证明式(i-a)、(i-b)发蓝绿色荧光,其发光峰位分别位于464nm和484nm,且经积分球测试其溶液发光效率分别为40.8%和53.2%,证明此类化合物可作为光致发光材料应用。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种制备1,2?二甲基咪唑的方法
- 一种芬苯达唑纳米混悬剂及其制备方法
- 吡啶并咪唑类衍生物、其制备方法和应用
- 一种咪唑类衍生物化合物及其制备方法和发光器件的制作方法
- Virosaine A的哌嗪基和咪唑基衍生物的组合物在抗红细胞低下性贫血药物中的应用
- Virosaine A的哌嗪基和咪唑基衍生物的组合物在抗骨质疏松药物中的应用
- Virosaine A的哌嗪基和咪唑基衍生物的组合物在抗急性痛风药物中的应用
- Virosaine A的哌嗪基和咪唑基衍生物的组合物在预防或治疗胰腺纤维化药物中的应用
- Virosaine A的哌嗪基和咪唑基衍生物的组合物在抗缺氧药物中的应用
- 适用于改进水分管理且提供抗微生物保护的包括衍生自直链四胺的季双咪唑啉化合物的 ...的制作方法