一种2-(四氢喹啉-6-基)-四氢-1,8-萘啶类化合物及其制备方法与应用与流程
本发明涉及医药化工合成技术领域,具体涉及一种2-(四氢喹啉-6-基)-四氢-1,8-萘啶类化合物及其制备方法与应用。
背景技术
近年来,杂环化合物,尤其是含氮的杂环化合物在现代药物研究中发挥着至关重要的作用,开发高效的含氮杂环化合物的构建方法也吸引了越来越多的关注。萘啶氮杂环化合物是一类极为重要的杂环体系,常见于药物中间体及多种天然产物中,具有潜在的生物活性。其中,四氢萘啶也是一种重要的杂环体系,在有机中间体、医药、农药、材料科学等领域都有广泛的应用前景。另外,四氢喹啉类有机化合物是一类较为常见的氮杂环化合物,其独特的分子结构大量存在于自然界中,它的各种衍生物大多都具有生物和药理活性。自然界中大部分具有生物活性的天然产物都含有一个或者多个氮杂环骨架。由单个杂环化合物组合形成新的双杂环或者多杂环结构时,往往会保留原有的生物活性,而且会产生意想不到的新特性。因此,促使氮杂环化合物之间的偶联,形成功能性更优越的双氮杂环化合物是当前研究的热点。
随着有机化学的发展以及材料、医药交叉学科发展,科学家们从含氮杂环化合物中发现了大量新的药物和新颖的功能材料,两个杂环以不同方式组合在一起的双氮杂环化合物在生物医药和功能材料方面扮演着十分重要的角色。四氢喹啉和萘啶结构是氮杂环化合物的基本骨架,许多天然产物的分子结构中都含有这些结构,并且有些化合物会表现出良好的生物活性。两个氮杂环化合物之间碳-碳键偶联一直是有机领域研究的热点和难点,结构比较复杂的双氮杂环骨架分子大都要经过许多步反应才能合成,不仅操作步骤极为繁琐,且原子和步骤经济性低,因此,在复杂双氮杂环的合成中,缩减合成路线是提高复杂氮杂环化合物最终产率的一种有效方式。然而,对于四氢喹啉与萘啶构建双氮杂环的合成方法,采用无金属催化目前为止并未有文献进行报道,基于此,若能探寻出一种无金属催化剂参与的、绿色的、高效的、操作简单的合成方法,将四氢喹啉和四氢萘啶结构单元之间进行有效组合,构建一系列含有潜在药物活性的双氮杂环骨架分子将具有重要意义。
技术实现要素:
本发明所要解决的技术问题是:提供一种对肿瘤具有抑制作用的2-(1,2,3,4-四氢喹啉-6-基)-1,2,3,4-四氢-1,8-萘啶类化合物及其制备方法与应用。
为了解决上述技术问题,本发明采用的技术方案为:一种2-(1,2,3,4-四氢喹啉-6-基)-1,2,3,4-四氢-1,8-萘啶类化合物,所述化合物的分子通式如下式i所示:
其中,所述式i中r1为苯基、硝基苯或卤代苯基;r2为氢或甲基;r3为氢或甲基;r4为氢或甲基。
本发明还包括上述2-(1,2,3,4-四氢喹啉-6-基)-1,2,3,4-四氢-1,8-萘啶类化合物的制备方法,包括以下步骤:
s1、在反应容器中,加入四氢喹啉类化合物和2-苯基-3-甲基-1,8萘啶类化合物后,加入四氢喹啉类化合物质量的1~5%的金属催化剂(catalyst,cat.),再加入酸和溶剂,在80℃~160℃下搅拌反应5~24小时,其中,所述四氢喹啉类化合物和1,8萘啶类化合物的摩尔比为1~2∶1,所述酸的质量为四氢喹啉类化合物质量的10~100%,其中,所述四氢喹啉类化合物优选为2-甲基-四氢喹啉或8-甲基-四氢喹啉,所述1,8萘啶类化合物优选为苯基被卤素或硝基取代后的2-苯基-3-甲基-1,8萘啶类化合物;
s2、反应结束后冷却至室温,过滤,减压旋蒸除去溶剂即可得到粗产物,粗产物经柱层析提纯得到2-四氢喹啉基-四氢萘啶类化合物。
进一步地,所述室温为20~30℃。
进一步地,所述步骤s1中,所述金属催化剂为醋酸铜、三氟甲磺酸铜、硫酸铜、氯化铜、氯化亚铜、氯化铁、醋酸钴、氯化钴、醋酸锰中的一种或两种以上的混合。
从上述描述可知,本发明的有益效果在于:采用廉价的金属催化剂即可实现有效的催化,大大降低了生产成本。
进一步地,所述步骤s1所述溶剂为乙醇、叔戊醇、异丙醇、1,4-二氧六环、n,n-二甲基甲酰胺、二甲基亚砜、甲苯、对二甲苯和水中的一种或两种以上的混合;所述溶剂的加入量与四氢喹啉类化合物的摩尔体积比优选为四氢喹啉类化合物:溶剂=0.5mmol:1~3ml。
进一步地,所述步骤s1所述酸为甲酸、乙酸、甲磺酸、苯甲酸、对甲苯磺酸、盐酸、三氟甲磺酸、三氟乙酸中的一种或两种以上的混合;所述酸与四氢喹啉类化合物的摩尔比优选为四氢喹啉类化合物:酸=0.5mmol:0.05mmol。
进一步地,所述步骤s1所述的反应容器优选为schlenk管(史兰克管),反应过程优选在氮气条件下进行。
进一步地,所述步骤s2所述柱层析提纯操作采用的洗脱液为体积比为
(0.5~50):(0~20):1的石油醚:二氯甲烷:乙酸乙酯的混合溶液。
其中,上述制备方法所涉及的部分反应方程式如下:
其中,所述的r1为苯基、硝基苯、卤素取代基苯;r2为氢或甲基;r3为氢或甲基;r4为氢或甲基。
本发明还包括上述2-(1,2,3,4-四氢喹啉-6-基)-1,2,3,4-四氢-1,8-萘啶类化合物在制备抗肿瘤试剂或防治肿瘤药物中的应用。
进一步地,所述应用为在抗人癌k562、hl-60、hela和/或bgc-823细胞的试剂或药物中。
本发明的有益效果在于:本发明提供了一种具有抗肿瘤活性的2-(1,2,3,4-四氢喹啉-6-基)-1,2,3,4-四氢-1,8-萘啶类化合物及其制备方法与应用,有效地解决了具有抗肿瘤活性的2-(1,2,3,4-四氢喹啉-6-基)-1,2,3,4-四氢-1,8-萘啶化合物的制备问题。通过本发明的制备方法可以高效合成2-(1,2,3,4-四氢喹啉-6-基)-1,2,3,4-四氢-1,8-萘啶类双氮杂环化合物,具有合成步骤简单、操作安全、原料无毒且原料价廉易得等优点,本发明制备过程选择性好且原子经济性高,得到的双氮杂环产物结构新颖,可进一步开发其生物活性;本发明的新型2-(1,2,3,4-四氢喹啉-6-基)-1,2,3,4-四氢-1,8-萘啶类化合物对受试人癌k562、hl-60、hela、bgc-823细胞具有良好的抑制作用,因此,该类化合物既作为抗肿瘤试剂用于体外抗肿瘤活性筛选中,也可用于防治肿瘤的药物中。
附图说明
图1为本发明实施例1制得的产物3a的氢谱图;
图2为本发明实施例1制得的产物3a的碳谱图;
图3为本发明实施例2制得的产物3b的氢谱图;
图4为本发明实施例2制得的产物3b的碳谱图;
图5为本发明实施例3制得的产物3c的氢谱图;
图6为本发明实施例3制得的产物3c的碳谱图;
图7为本发明实施例4制得的产物3d的氢谱图;
图8为本发明实施例4制得的产物3d的碳谱图;
图9为本发明实施例5制得的产物3e的氢谱图;
图10为本发明实施例5制得的产物3e的碳谱图。
具体实施方式
为详细说明本发明的技术内容、所实现目的及效果,以下结合实施方式并配合附图予以说明。
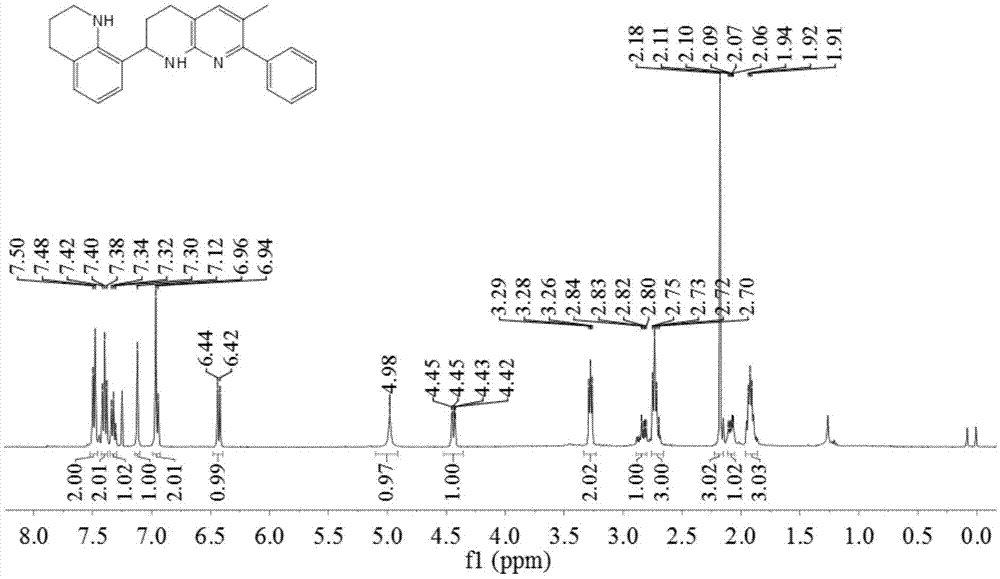
本发明的实施例1为一种具有抗肿瘤活性的2-(1,2,3,4-四氢喹啉-6-基)-1,2,3,4-四氢-1,8-萘啶的制备方法,包括以下步骤:在反应器中,加入1.0毫摩尔四氢喹啉和0.5毫摩尔2-苯基-3-甲基-1,8-萘啶,加入1%的三氟甲磺酸铜(catalyst,cat.),再加入50%的三氟甲磺酸,1.5ml甲苯,在80℃下搅拌反应5小时,反应结束后冷却至室温,稀释反应液,过滤,减压旋蒸除去溶剂即可得到粗产物,粗产物经柱层析提纯得到3a,该产物呈黄色油状。
所得产物3a的氢谱图和碳谱图分别如图1和图2所示,结构表征数据如下:
1hnmr(400mhz,cdcl3):
δ7.49(d,j=7.5hz,2h),7.40(t,j=7.5hz,2h),7.32(t,j=7.2hz,1h),7.12(s,1h),6.95(d,j=7.7hz,2h),6.43(d,j=7.8hz,1h),4.98(s,1h),4.44(dd,j=9.1,2.8hz,1h),3.33-3.23(m,2h),2.89-2.80(m,1h),2.76-2.66(m,3h),2.18(s,3h),2.11-2.05(m,1h),1.96-1.86(m,3h);
13cnmr(101mhz,cdcl3):
δ154.28,144.15,141.15,139.07,132.32,128.95,128.02,127.43,127.35,124.77,121.45,118.86,114.72,114.18,55.79,42.02,30.92,27.04,25.53,22.20,18.77;
将所得产物3a通过红外光谱仪(infraredspectrometer,ir(kbr))测定其红外吸收光谱结果发现,该化合物在2928、1588、1452、1308、1131、819、750、699cm-1处有特征吸收峰,表明了本发明方案制得了带有目标官能团的产物。
通过电喷雾电离源(electronsprayionization,esi)的高分辨质谱仪(highresolutionmassspectrometer,hrms)测定该化合物的分子量,c24h26n3[m+h]+:理论值(calculatedvalue,calcd.):356.2121;实际测得值(found):356.2125。
本发明实施例2为一种具有抗肿瘤活性的2-(1,2,3,4-四氢喹啉-6-基)-1,2,3,4-四氢-1,8-萘啶类化合物的制备方法,包括以下步骤:在反应器中,加入0.8毫摩尔2-甲基四氢喹啉和0.5毫摩尔2-苯基-3-甲基-1,8-萘啶,加入2-甲基四氢喹啉质量5%的氯化铁,再加入60%的甲磺酸,1.0ml甲苯,在160℃下搅拌反应12小时,反应结束后冷却至室温,稀释反应液,过滤,减压旋蒸除去溶剂即可得到粗产物,粗产物经柱层析提纯得到产物3b,所述产物3b呈黄色油状。
所得产物3b的氢谱图和碳谱图分别如图3和图4所示,结构表征数据如下:
1hnmr(400mhz,cdcl3)δ7.87(d,j=4.8hz,1h),7.18(d,j=7.1hz,1h),6.95(d,j=9.8hz,2h),6.54-6.47(m,1h),6.44(d,j=7.9hz,1h),5.04(s,1h),4.43(dd,j=9.0,2.8hz,1h),3.55-3.28(m,1h),2.88-2.77(m,2h),2.72-2.64(m,2h),2.11-2.02(m,1h),1.97-1.82(m,2h),1.65-1.53(m,1h),1.21(d,j=6.2hz,3h);
13cnmr(101mhz,cdcl3)δ156.41,145.85,144.28,136.02,132.11,127.20,124.78,121.15,115.94,113.99,112.70,55.72,47.21,30.54,30.10,26.63,25.84,22.59;
ir(kbr)结果表明,产物3b在2936、2845、1860、1819、1489、1352、1330、1292、1194、1170、771、739cm-1均有特征吸收峰,该产物带有目标官能团。
通过电喷雾电离源(electronsprayionization,esi)的高分辨质谱仪(highresolutionmassspectrometer,hrms)测定该化合物的分子量,c18h22n3[m+h]+理论值(calcd.):280.1808;实际测得值(found):280.1804。
本发明的实施例3为一种具有抗肿瘤活性的2-(1,2,3,4-四氢喹啉-6-基)-1,2,3,4-四氢-1,8-萘啶类化合物的制备方法,包括以下步骤:在反应器中,加入0.75毫摩尔2-甲基四氢喹啉和0.5毫摩尔2-(3-硝基苯基)-1,8-萘啶,加入2-甲基四氢喹啉2%的醋酸钴催化剂,再加入10%的对甲苯磺酸,1.2ml甲苯,在130℃下搅拌反应15小时,反应结束后冷却至室温,稀释反应液,过滤,减压旋蒸除去溶剂即可得到粗产物,粗产物经柱层析提纯得到产物3c,所述产物3c呈黄色油状。
所得产物3c的氢谱图和碳谱图分别如图5和图6所示,结构表征数据如下:
1hnmr(400mhz,cdcl3)δ8.80(s,1h),8.27(d,j=7.8hz,1h),8.16(d,j=8.1hz,1h),7.56(d,j=7.9hz,1h),7.30(d,j=7.4hz,1h),7.03(d,j=7.4hz,1h),6.96(d,j=12.5hz,2h),6.45(d,j=8.0hz,1h),5.19(s,1h),4.47(d,j=7.5hz,1h),3.72(s,1h),3.44-3.37(m,1h),2.89-2.70(m,4h),2.11(dd,j=12.9,3.9hz,1h),2.00-1.91(m,2h),1.64-1.53(m,1h),1.21(d,j=6.2hz,3h);
13cnmr(101mhz,cdcl3)δ156.39,151.04,148.69,144.37,141.66,136.77,132.30,131.79,129.29,127.22,124.82,122.68,121.47,121.19,116.17,114.00,109.58,55.80,47.23,30.42,30.12,26.66,25.70,22.59;
ir(kbr)结果表明,产物3c在3415、1638、1618、1527、1442、1348、1121、870、792、734cm-1均有特征吸收峰,该产物带有目标官能团。
通过电喷雾电离源(electronsprayionization,esi)的高分辨质谱仪(highresolutionmassspectrometer,hrms)测定该化合物的分子量,calcd.forc24h25n4o2[m+h]+:401.1972;found:401.1975。
本发明的实施例4为一种具有抗肿瘤活性的2-(1,2,3,4-四氢喹啉-6-基)-1,2,3,4-四氢-1,8-萘啶类化合物的制备方法,包括以下步骤:在反应器中,加入0.8毫摩尔8-甲基四氢喹啉和0.4毫摩尔2-(4-氯苯基)-1,8-萘啶,加入8-甲基四氢喹啉3%的醋酸铜催化剂,再加入50%的三氟乙酸,1.2ml叔戊醇,在100℃下搅拌反应10小时,反应结束后冷却至室温,稀释反应液,过滤,减压旋蒸除去溶剂即可得到粗产物,粗产物经柱层析提纯得到产物3d,所述产物3d呈棕色固体状,熔点为140-141℃。
所得产物3d的氢谱图和碳谱图分别如图7和图8所示,结构表征数据如下:
1hnmr(400mhz,cdcl3)δ7.86(d,j=8.5hz,2h),7.36(d,j=8.5hz,2h),7.25(t,j=3.7hz,1h),6.93(d,j=7.5hz,1h),6.88(d,j=10.2hz,2h),5.15(s,1h),4.44(dd,j=9.1,3.1hz,1h),3.40-3.30(m,2h),2.87-2.71(m,4h),2.12-2.07(m,1h),2.07(s,3h),1.96-1.88(m,3h);
13cnmr(101mhz,cdcl3)δ156.18,152.54,142.20,138.28,136.76,134.12,131.35,128.61,127.87,125.96,125.34,121.33,120.91,115.09,109.37,55.89,42.36,30.75,27.36,25.81,22.14,17.22;
ir(kbr)结果表明,产物3d在2928、2857、1665、1627、1594、1462、1304、1088、806、752cm-1均有特征吸收峰,该产物带有目标官能团。
通过电喷雾电离源(electronsprayionization,esi)的高分辨质谱仪(highresolutionmassspectrometer,hrms)测定该化合物的分子量,calcd.forc24h25cln3[m+h]+:390.1731;found:390.1735.
本发明的实施例5为一种具有抗肿瘤活性的2-(1,2,3,4-四氢喹啉-6-基)-1,2,3,4-四氢-1,8-萘啶类化合物的制备方法,包括以下步骤:在反应器中,加入0.4毫摩尔8-甲基四氢喹啉和0.4毫摩尔2-(4-溴苯基)-1,8-萘啶,加入8-甲基四氢喹啉质量3%的氯化铜催化剂,再加入80%的对甲苯磺酸,1.2ml对二甲苯,在150℃下搅拌反应10小时,反应结束后冷却至室温,稀释反应液,过滤,减压旋蒸除去溶剂即可得到粗产物,粗产物经柱层析提纯得到3e,所述产物3e呈黄色固体状,熔点为173-174℃。
所得产物3e的氢谱图和碳谱图分别如图9和图10所示,结构表征数据如下:
1hnmr(400mhz,cdcl3)δ7.84(d,j=8.4hz,2h),7.55(d,j=8.4hz,2h),7.32-7.27(m,1h),6.97(d,j=7.5hz,1h),6.92(d,j=10.3hz,2h),5.18(s,1h),4.48(dd,j=9.2,3.0hz,1h),3.46-3.36(m,2h),2.90-2.75(m,4h),2.18-2.11(m,1h),2.11(s,3h),1.99-1.88(m,3h);
13cnmr(101mhz,cdcl3)δ156.21,142.20,136.76,131.56,131.34,128.19,125.96,125.34,122.42,121.33,120.90,115.17,109.34,55.89,42.36,30.75,27.37,25.83,22.14,17.22;
ir(kbr)结果表明,产物3e在2928、1633、1591、1501、1462、1321、807cm-1均有特征吸收峰,该产物带有目标官能团。
通过电喷雾电离源(electronsprayionization,esi)的高分辨质谱仪(highresolutionmassspectrometer,hrms)测定该化合物的分子量,calcd.forc24h25brn3[m+h]+:434.1226;found:434.1224。
将上述实施例1-5所制得的新化合物3a-3e,进行抗肿瘤活性测试:将各个化合物分别配制成100μg·ml-1的甲醇溶液,阳性对照药5-氟尿嘧啶(5-fluorouracil,5-fu)和多烯紫杉醇(docetaxol)分别配成100μg·ml-1的二甲基亚砜(dimethylsulfoxide,dmso)溶液,分别以甲醇和dmso溶剂为空白对照,采用四甲基偶氮唑盐(methylthiazolyltetrazoliummethod,mtt)法测试各化合物对k562、hl-60、hela、bgc-823细胞的抑制作用,具体操作如下:
1、细胞培养液的配制:取一袋rpmi-1640培养基粉末(净含量为10.4g)倒入干净的烧杯中,用900ml超净水将其溶解,并加入100mg·ml-1的链霉素1ml、青霉素0.5ml及nahco32g。磁力搅拌均匀后,于超净台中用已高压灭菌的蔡氏(zeiss)滤器经0.22μm滤膜过滤除菌,滤液直接保存于湿热灭菌后的玻璃瓶中(450ml/瓶)。培养基使用前,取冷冻保藏的血清,56℃灭活30min后,加入已配好rpmi-1640培养液中(450ml培养基中加入50ml血清),轻轻摇匀后,加盖,用锡箔纸封口,于4℃冰箱中保存。
mtt溶液配制:50mg的mtt粉末溶解于10ml的pbs溶液中,用0.22μm的滤膜过滤,于4℃冰箱中保存。
2、抗肿瘤活性实验:分别取生长对数期的k562、hl-60、hela、bgc-823细胞,于4℃、3000rpm离心机上离心3min,吸去上清液,加入新鲜的rpmi-1640培养基稀释成1×105个/毫升的细胞悬液。每孔200μl接种于96孔板中,于37℃、5%co2的细胞培养箱中培养1h后,每孔各加样品溶液2μl,每个样品设3个平行孔,另设两组各三孔的空白对照,加样后以相同条件培养24h。24h后,在光学显微镜下观察细胞有无形态变化,初步判断样品有无细胞毒活性,必要时进行拍照。每孔加入5mg·ml-1的mtt溶液各20μl,培养箱中继续培养4h。取出96孔板离心(4℃、2000rpm,20min)除去上清液,每孔各加150μldmso,充分振荡使紫色沉淀物完全溶解。在酶标仪上于570nm下测定其光密度od值,每组样品取平均值并按抑制率(inhibitionrate,ir)%=(od空白-od样品)/od空白×100%公式计算。
mtt法测试五种2-(1,2,3,4-四氢喹啉-6-基)-1,2,3,4-四氢-1,8-萘啶类双氮杂环类化合物对四种肿瘤细胞的增殖活性抑制结果如表1所示:
表1mtt法测试类化合物对四种肿瘤细胞的增殖活性抑制作用的结果
由上表1可以看出,本发明方案制得的化合物对k562、hl-60、hela、bgc-823细胞均具有一定的抑制作用。
综上所述,本发明提供的一种2-(四氢喹啉-6-基)-四氢-1,8-萘啶类化合物及其制备方法与应用,该类化合物对肿瘤具有抑制作用,本发明方案的制备方案绿色环保且经济效益好,具有良好的生产应用前景。
以上所述仅为本发明的实施例,并非因此限制本发明的专利范围,凡是利用本发明说明书及附图内容所作的等同变换,或直接或间接运用在相关的技术领域,均同理包括在本发明的专利保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!