一种潜伏型光敏微胶囊环氧固化剂及其制备方法与流程
本发明涉及胶粘剂技术领域,具体涉及一种潜伏型光敏微胶囊环氧固化剂及其制备方法。
背景技术:
环氧树脂是泛指分子中含有两个或两个以上环氧基团的低分子质量有机化合物,在常温和一般加热条件下不会固化,由于其分子结构中含有活泼的环氧基团,可用环氧固化剂使其开环,形成不溶的具有网状结构的高聚物。固化后的环氧树脂具有良好的物理、化学性能,如它对各种材料的黏着力很强、固化收缩率小、电绝缘性好、有很强的耐化学腐蚀性等,广泛应用于涂料、胶粘剂、玻璃钢、层压板、电子浇铸、灌封、包封等领域。
在环氧树脂胶粘剂应用中,通常胶粘剂是以环氧树脂组分和固化剂分开的双组分包装形式提供应用。但是双组分混合给使用带来不方便,例如增加了包装和贮运的麻烦;双组分胶粘剂使用时,混合比例的准确性和均一性将影响粘接强度;在树脂和固化剂混合后使用时间短等。而单组分胶粘剂避免了上述缺点,一般人们将双氰胺、bf3-胺络合物、胺盐、改性咪唑化合物等潜在性固化剂混合到环氧树脂中而得到的组合物,这可以使胶的粘接工艺简化,并适于自动化操作,便于现代大规模工业化生产。目前国内外市场出售的单组分环氧树脂胶粘剂几乎都是采用潜伏性固化剂,产品的形态有液态、糊状、粉末状和膜状。但是,人们在使用这些单组分环氧树脂胶粘剂时发现,储藏稳定性优异的胶粘剂的固化性较低,固化时需要高温或长时间,而固化性高的胶粘剂由于活性高其储藏稳定性较低。对于这些问题,人们利用微胶囊技术将固化剂用以特定的壳包覆起来,阻止其与基体环氧树脂反应,在使用时通过加热或加压等,使微胶囊破裂,释放出固化剂,完成与基体环氧树脂的固化反应,这基本解决了环氧树脂低温固化和较长室温储存期之间的矛盾。但是目前在利用微胶囊潜在固化剂进行环氧树脂的固化反应中,由于固化时微胶囊破裂不完全,胶囊破裂后释放固化剂不充分等原因导致了体系反应所需要的时间较长、快速固化性不充分等问题。
技术实现要素:
本发明要解决现有技术中的技术问题,提供一种潜伏型光敏微胶囊环氧固化剂及其制备方法。
为了解决上述技术问题,本发明的技术方案具体如下:
一种潜伏型光敏微胶囊环氧固化剂,包括微胶囊壁材料和环氧固化剂;所述微胶囊壁材料用于包裹所述环氧固化剂;
其特征在于:
所述微胶囊壁材料为结构通式为a3、b3或者c3的聚合物:
式中:
r为甲基丙烯酸酯类单体、丙烯酸酯类单体、苯乙烯单体、苯乙烯衍生物单体、甲基丙烯酯类单体和丙烯酯类单体中的一种;左右两端的r可以相同,也可以不同;
m+n为1-20000;
或者所述微胶囊壁材料为上述结构通式所示的聚合物和其它高分子材料的混合物。
在上述技术方案中,r为甲基丙烯酸甲酯单体、丙烯酸甲酯单体或苯乙烯单体。
在上述技术方案中,所述潜伏型光敏微胶囊环氧固化剂的粒径为1-100μm。
在上述技术方案中,所述潜伏型光敏微胶囊环氧固化剂的粒径为5-30μm。
在上述技术方案中,所述聚合物和其它高分子材料的混合物中聚合物的掺杂比例大于90%。
一种潜伏型光敏微胶囊环氧固化剂的制备方法,包括以下步骤:
步骤1、结构通式为a3、b3或者c3的聚合物的制备
步骤1-1、将3-羟甲基-2硝基苄基醇a1或者4-羟甲基-2硝基苄基醇b1或者5-羟甲基-2硝基苄基醇c1溶解在干燥二氯甲烷或氯仿溶剂制成质量分数为0.1-10%的溶液,将该溶液加入到装有搅拌器的三口瓶中,再加入4-二甲基氨基吡啶催化剂,搅拌均匀后,加入三乙胺后,滴加2-溴-2-甲基-丙酰溴,室温搅拌,反应20-30小时,用甲醇淬灭反应,四氢呋喃溶剂稀释反应液,用200目-500目的碱性硅胶或碱性三氧化二铝过滤三乙胺盐酸盐,将滤液滴入乙醚中,析出沉淀,用布氏漏斗过滤,再多次用去离子水洗,100~110℃真空干燥10~15h得到中间体a2或者b2或者c2;
所述4-二甲基氨基吡啶的质量为3-羟甲基-2硝基苄基醇a1或者4-羟甲基-2硝基苄基醇b1或者5-羟甲基-2硝基苄基醇c1质量的0.01-0.02倍;
所述三乙胺的质量为3-羟甲基-2硝基苄基醇a1或者4-羟甲基-2硝基苄基醇b1或者5-羟甲基-2硝基苄基醇c1质量的0.04-0.06倍;
所述2-溴-2-甲基-丙酰溴的摩尔数为3-羟甲基-2硝基苄基醇a1或者4-羟甲基-2硝基苄基醇b1或者5-羟甲基-2硝基苄基醇c1摩尔数的2-2.2倍;
所述甲醇的质量为3-羟甲基-2硝基苄基醇a1或者4-羟甲基-2硝基苄基醇b1或者5-羟甲基-2硝基苄基醇c1质量的0.1-0.2倍;
步骤1-2、在氮气气氛下向装有搅拌器的施兰克反应瓶中,加入中间体a2或者中间体b2或者中间体c2,和溴化亚铜,再加入可以进行原子转移自由基聚合反应的r单体,再滴加n,n,n',n”,n”-五甲基二亚乙基三胺,经过3次冷冻、抽真空和通氮气解冻后,在真空条件下封管,在油浴中将反应液加热到90-100℃,反应8-24小时,体系放凉后,将反应混合物倒入水中,得到聚合物,再用粉碎机粉碎成细颗粒,然后用布氏漏斗过滤,再多次用去离子水洗,乙醇洗后100~110℃真空干燥10~15h,得到产物;
所述溴化亚铜的摩尔数是中间体a2或者中间体b2或者中间体c2摩尔数的1-1.1倍;
所述n,n,n',n”,n”-五甲基二亚乙基三胺的摩尔数是中间体a2或者中间体b2或者中间体c2摩尔数的1-1.1倍;
所述r单体为甲基丙烯酸酯类单体、丙烯酸酯类单体、苯乙烯单体、苯乙烯衍生物单体、甲基丙烯酯类单体和丙烯酯类单体中的一种;式a3、b3、c3中左右两端的r可以相同,也可以不同;m+n为1-20000;
合成路线如下:
步骤2、潜伏型光敏微胶囊环氧固化剂的制备
步骤2-1、环氧固化剂溶液的配制
将环氧固化剂配置成质量分数在0.01-10%的溶液;
具体的操作:
将环氧固化剂逐渐加入到装有溶剂的反应釜中,加热到30-90℃,用搅拌桨搅拌5-60分钟,至环氧固化剂完全溶解;
步骤2-2、微胶囊壁材料的溶解
将步骤1制备的聚合物、或者该聚合物和其它高分子材料的混合物,加入到步骤1-1配制的环氧固化剂溶液中,加热到30-90℃,用搅拌桨搅拌30-60分钟,至完全溶解;
步骤2-3、表面活性剂水溶液的配制
向另外的反应釜中,加入去离子水,并按加入水量的质量份数1.5%-3.5%的比例加入表面活性剂,在搅拌条件下使其完全溶解;
步骤2-4、微胶囊环氧固化剂的制备
在机械搅拌条件下,将步骤2-2配制的微胶囊环氧固化剂溶液逐滴滴加到步骤2-3配制的表面活性剂水溶液中,一边滴加一边搅拌,搅拌速度为500-1000r/min,保持4-9小时,待溶剂完全挥发后,停止搅拌;
步骤2-5、微胶囊环氧固化剂的分离提纯
将步骤2-4得到的微胶囊环氧固化剂悬浊液,在3000-6000r/min的速度下离心,得到下层浑浊液用无水乙醇超声洗涤20-60分钟,超声结束后,将烧杯中的固体物质用无水乙醇反复洗涤2-3次,过滤后,将固体置于50℃烘箱中2-9小时烘干,最后得到干燥的固体粉末,即微胶囊环氧固化剂。
在上述技术方案中,步骤2-1中所述环氧固化剂为邻苯二甲酸酐、六氢邻苯二甲酸酐、四氢邻苯二甲酸酐、丙二醇改性聚硫醇、三羟甲基丙烷的硫代葡糖酸酯、聚硫树脂、三氟硼的乙胺盐、1,8-二氮杂双环[5.4.0]-十一-7-烯的苯酚盐、3-苯基-1,1-二甲基脲、三苯基膦或四苯基膦四苯基硼酸酯;
或者为间苯二甲胺、二氨基二苯基甲烷、氨基二苯基砜、间苯二胺和n-氨乙基哌嗪中的一种或几种的混合胺类固化剂。
在上述技术方案中,步骤2-1中所述溶剂为二氯甲烷、氯仿、四氯化碳、乙酸乙酯、苯、甲苯或者二甲苯。
在上述技术方案中,步骤2-1中所述搅拌桨为框式搅拌桨或锚式搅拌桨。
在上述技术方案中,步骤2-3中所述表面活性剂为十二烷基硫酸钠、硬脂酸聚氧乙烯酯、丙烯酸二乙胺基酯/丙烯酰胺共聚物、二甲胺基甲基丙烯酰胺/丙烯酸共聚物或明胶。
本发明具有以下的有益效果:
本发明的微胶囊环氧固化剂是一种以新型结构的可降解的聚合物作为微胶囊壁材料、以环氧固化剂为芯材料的微胶囊环氧固化剂。本发明的微胶囊环氧固化剂的微胶囊壁材料能在200-400nm波长下降解,进而释放环氧固化剂用于固化环氧树脂。解决了目前在利用微胶囊潜在固化剂进行环氧树脂的固化反应中,由于固化时微胶囊破裂不完全,胶囊破裂后释放固化剂不充分等原因导致了体系反应所需要的时间较长、快速固化性不充分等问题。
本发明借助乳液-溶剂蒸发技术用该聚合物或该聚合物掺杂其它高分子材料的混合物为微胶囊壁材料,包裹环氧树脂固化剂,制备成粒径为1-100μm圆形微胶囊固化剂。并利用该种微胶囊壁材料能在200-400nm波长下可降解的能力,释放环氧固化剂固化环氧树脂。
附图说明
下面结合附图和具体实施方式对本发明作进一步详细说明。
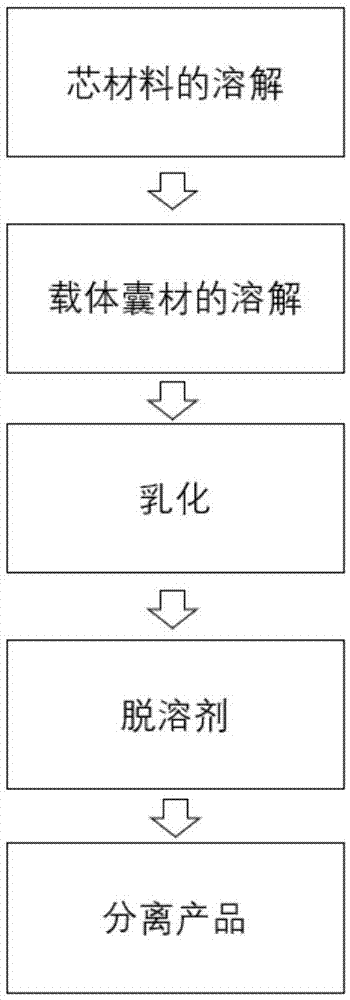
图1为本发明的微胶囊环氧固化剂的制备流程图。
图2为本发明实施例1制备的微胶囊环氧固化剂的扫描电子显微镜照片。
图3为本发明实施例2制备的微胶囊环氧固化剂的扫描电子显微镜照片。
图4为本发明实施例3制备的微胶囊环氧固化剂的扫描电子显微镜照片。
图5为本发明实施例4制备的微胶囊环氧固化剂的扫描电子显微镜照片。
图6为本发明实施例5制备的微胶囊环氧固化剂的扫描电子显微镜照片。
图7为本发明实施例6制备的微胶囊环氧固化剂的扫描电子显微镜照片。
图8为本发明实施例6制备的微胶囊环氧固化剂的释放率随在紫外光的照射时间变化曲线。
具体实施方式
下面结合附图对本发明做以详细说明。
本发明提供一种潜伏型光敏微胶囊环氧固化剂,包括微胶囊壁材料和环氧固化剂;所述微胶囊壁材料用于包裹所述环氧固化剂;
所述微胶囊壁材料为结构通式为a3、b3或者c3的聚合物:
式中:r为甲基丙烯酸酯类单体、丙烯酸酯类单体、苯乙烯单体、苯乙烯衍生物单体、甲基丙烯酯类单体和丙烯酯类单体中的一种;左右两端的r可以相同,也可以不同;m+n为1-20000;或者所述微胶囊壁材料为上述结构通式所示的聚合物和其它高分子材料的混合物。优选r为甲基丙烯酸甲酯单体、丙烯酸甲酯单体或苯乙烯单体。优选所述潜伏型光敏微胶囊环氧固化剂的粒径为1-100μm。再优选所述潜伏型光敏微胶囊环氧固化剂的粒径为5-30μm。优选所述聚合物和其它高分子材料的混合物中聚合物的掺杂比例大于90%。
结合图1说明本发明还提供一种潜伏型光敏微胶囊环氧固化剂的制备方法,包括以下步骤:
步骤1、结构通式为a3、b3或者c3的聚合物的制备
步骤1-1、将3-羟甲基-2硝基苄基醇a1或者4-羟甲基-2硝基苄基醇b1或者5-羟甲基-2硝基苄基醇c1溶解在干燥二氯甲烷或氯仿溶剂制成质量分数为0.1-10%的溶液,将该溶液加入到装有搅拌器的三口瓶中,再加入4-二甲基氨基吡啶催化剂,搅拌均匀后,加入三乙胺后,滴加2-溴-2-甲基-丙酰溴,室温搅拌,反应20-30小时,用甲醇淬灭反应,四氢呋喃溶剂稀释反应液,用200目-500目的碱性硅胶或碱性三氧化二铝过滤三乙胺盐酸盐,将滤液滴入乙醚中,析出沉淀,用布氏漏斗过滤,再多次用去离子水洗,100~110℃真空干燥10~15h得到中间体a2或者b2或者c2;
所述4-二甲基氨基吡啶的质量为3-羟甲基-2硝基苄基醇a1或者4-羟甲基-2硝基苄基醇b1或者5-羟甲基-2硝基苄基醇c1质量的0.01-0.02倍;
所述三乙胺的质量为3-羟甲基-2硝基苄基醇a1或者4-羟甲基-2硝基苄基醇b1或者5-羟甲基-2硝基苄基醇c1质量的0.04-0.06倍;
所述2-溴-2-甲基-丙酰溴的摩尔数为3-羟甲基-2硝基苄基醇a1或者4-羟甲基-2硝基苄基醇b1或者5-羟甲基-2硝基苄基醇c1摩尔数的2-2.2倍;
所述甲醇的质量为3-羟甲基-2硝基苄基醇a1或者4-羟甲基-2硝基苄基醇b1或者5-羟甲基-2硝基苄基醇c1质量的0.1-0.2倍;
步骤1-2、在氮气气氛下向装有搅拌器的施兰克反应瓶中,加入中间体a2或者中间体b2或者中间体c2,和溴化亚铜,再加入可以进行原子转移自由基聚合反应的r单体,再滴加n,n,n',n”,n”-五甲基二亚乙基三胺,经过3次冷冻、抽真空和通氮气解冻后,在真空条件下封管,在油浴中将反应液加热到90-100℃,反应8-24小时,体系放凉后,将反应混合物倒入水中,得到聚合物,再用粉碎机粉碎成细颗粒,然后用布氏漏斗过滤,再多次用去离子水洗,乙醇洗后100~110℃真空干燥10~15h,得到产物;
所述溴化亚铜的摩尔数是中间体a2或者中间体b2或者中间体c2摩尔数的1-1.1倍;
所述n,n,n',n”,n”-五甲基二亚乙基三胺的摩尔数是中间体a2或者中间体b2或者中间体c2摩尔数的1-1.1倍;
所述r单体为甲基丙烯酸酯类单体、丙烯酸酯类单体、苯乙烯单体、苯乙烯衍生物单体、甲基丙烯酯类单体和丙烯酯类单体中的一种;式a3、b3、c3中左右两端的r可以相同,也可以不同;m+n为1-20000;
合成路线如下:
步骤2、潜伏型光敏微胶囊环氧固化剂的制备
步骤2-1、环氧固化剂溶液的配制(芯材料的溶解)
将环氧固化剂配置成质量分数在0.01-10%的溶液,具体的操作就是称取一定质量的环氧固化剂,逐渐加入适量溶剂的反应釜中,加热到30-90℃,用特定的搅拌桨搅拌5-60分钟,待固化剂完全溶解;
所述环氧固化剂为邻苯二甲酸酐、六氢邻苯二甲酸酐、四氢邻苯二甲酸酐、丙二醇改性聚硫醇、三羟甲基丙烷的硫代葡糖酸酯、聚硫树脂、三氟硼的乙胺盐、1,8-二氮杂双环[5.4.0]-十一-7-烯的苯酚盐、3-苯基-1,1-二甲基脲、三苯基膦或四苯基膦四苯基硼酸酯;或者为间苯二甲胺、二氨基二苯基甲烷、氨基二苯基砜、间苯二胺和n-氨乙基哌嗪中的一种或几种的混合胺类固化剂。所述溶剂为二氯甲烷、氯仿、四氯化碳、乙酸乙酯、苯、甲苯或者二甲苯。所述搅拌桨为框式搅拌桨或锚式搅拌桨;
步骤2-2、微胶囊壁材料的溶解(载体囊材的溶解)
将步骤1制备的聚合物、或者该聚合物和其它高分子材料的混合物,加入到步骤1-1配制的环氧固化剂溶液中,加热到30-90℃,用搅拌桨搅拌30-60分钟,待该聚合物完全溶解,根据聚合物溶解性,选择合适的溶剂增溶,例如,甲苯、二甲苯、苯、氯苯等溶剂。
步骤2-3、表面活性剂水溶液的配制(乳化过程)
向另外的反应釜中,加入适量的去离子水,并按加入水量的质量份数1.5%-3.5%的比例加入表面活性剂,在搅拌条件下使其完全溶解;
所述表面活性剂为十二烷基硫酸钠、硬脂酸聚氧乙烯酯、丙烯酸二乙胺基酯/丙烯酰胺共聚物、二甲胺基甲基丙烯酰胺/丙烯酸共聚物或明胶。
步骤2-4、微胶囊环氧固化剂的制备(脱溶剂过程)
在机械搅拌条件下,将步骤2-2配制的微胶囊环氧固化剂溶液逐滴滴加到步骤2-3配制的表面活性剂水溶液中,一边滴加一边搅拌,搅拌速度为500-1000r/min,保持4-9小时,待溶剂完全挥发后,停止搅拌;
步骤2-5、微胶囊环氧固化剂的分离提纯(产品分离)
将步骤2-4得到的微胶囊环氧固化剂悬浊液,在3000-6000r/min的速度下离心,得到下层浑浊液用无水乙醇超声洗涤20-60分钟,超声结束后,将烧杯中的固体物质用无水乙醇反复洗涤2-3次,过滤后,将固体置于50℃烘箱中2-9小时烘干,最后得到干燥的固体粉末,即微胶囊环氧固化剂。
实施例1
微胶囊壁材料结构如下图所示,
聚合物a-1的制备
在装有搅拌器的三口瓶中,将183mg3-羟甲基-2硝基苄基醇(a1)溶解在90ml干燥二氯甲烷中,再加入1.83mg4-二甲基氨基吡啶。搅拌均匀后,加入3mg三乙胺后,滴加459.8mg2-溴-2-甲基-丙酰溴,室温搅拌,反应20小时,用18ml甲醇淬灭反应。向反应液中加入10ml四氢呋喃溶剂,用200目碱性三氧化二铝过滤三乙胺盐酸盐,将滤液滴入乙醚中,析出沉淀,用布氏漏斗过滤,再多次用去离子水洗,110℃真空干燥10h得到394mg中间体a2,产率,82%。
质谱分析,[m+h]+:479.96;元素分析,c39.93h3.97br33.20n2.90o19.94。红外(cm-1):3003,2980,2933,1739,1534,1464,1363,1271,1154,1109。
在氮气气氛下向装有搅拌器的施兰克反应瓶中,加入481mg中间体a2和143mg溴化亚铜,加入10ml甲基丙烯酸甲酯(pmma),再滴加173mgn,n,n',n”,n”-五甲基二亚乙基三胺经过3次冷冻—抽真空—通氮气解冻后,在真空条件下封管,在油浴中将反应液加热到100℃,反应24小时。体系放凉后,将反应混合物倒入水中,得到聚合物,再用粉碎机粉碎成细颗粒,然后用布氏漏斗过滤,再多次用去离子水洗,乙醇洗后110℃真空干燥15h,得到产物聚合物a-1。
红外(cm-1):2977,2933,2879,1730,1540,1448,1394,1369,1258,1154,846,736。分子量:mn=39800,pdi=1.10。
微胶囊固化剂的制备
称取10g间苯二胺,逐渐加入到含有1000g氯仿的反应釜1中,加热到40℃,用锚式桨搅拌50分钟,待固化剂完全溶解。称取40g聚合物a-1将其加入已配制好的固化剂溶液中,加热到40℃,用锚式搅拌桨搅拌45分钟。
另外的反应釜2中,加入1000g去离子水,并按加入水量的质量份数15g十二烷基硫酸钠,在搅拌条件下使其完全溶解;在机械搅拌条件下,将反应釜1配制好的溶液逐滴滴加进反应釜2配制好的水溶液中;滴加完成后升高搅拌速度至1000r/min,保持9小时;待溶剂完全挥发后,停止搅拌。
将悬浊液,在6000r/min的速度下离心,得到下层浑浊液用无水乙醇超声洗涤20分钟,超声结束后,将烧杯中的固体物质用无水乙醇反复洗涤2-3次,过滤后,将固体置于50℃烘箱中8小时烘干,最后得到干燥的固体粉末,即微胶囊固化剂。
实施例2
微胶囊壁材料
聚合物a-2的制备
在装有搅拌器的三口瓶中,将183mg3-羟甲基-2硝基苄基醇(a1)溶解在90ml干燥二氯甲烷中,再加入1.83mg4-二甲基氨基吡啶。搅拌均匀后,加入3mg三乙胺后,滴加459.8mg2-溴-2-甲基-丙酰溴,室温搅拌,反应20小时,用18ml甲醇淬灭反应。向反应液中加入10ml四氢呋喃溶剂,用200目碱性三氧化二铝过滤三乙胺盐酸盐,将滤液滴入乙醚中,析出沉淀,用布氏漏斗过滤,再多次用去离子水洗,110℃真空干燥10h得到394mg中间体a2,产率,82%。
质谱分析,[m+h]+:479.96;元素分析,c39.93h3.97br33.20n2.90o19.94。红外(cm-1):3003,2980,2933,1739,1534,1464,1363,1271,1154,1109。
在氮气气氛下向装有搅拌器的施兰克反应瓶中,加入481mg中间体a2和143mg溴化亚铜,加入5ml甲基丙烯酸甲酯(pmma)和5ml苯乙烯(ps),再滴加173mgn,n,n',n”,n”-五甲基二亚乙基三胺经过3次冷冻—抽真空—通氮气解冻后,在真空条件下封管,在油浴中将反应液加热到100℃,反应24小时。体系放凉后,将反应混合物倒入水中,得到聚合物,再用粉碎机粉碎成细颗粒,然后用布氏漏斗过滤,再多次用去离子水洗,乙醇洗后110℃真空干燥15h,得到产物聚合物a-2。
红外(cm-1):2977,2933,2880,1731,1528,1445,1384,1369,1258,1154,846,736。分子量:mn=39100,pdi=1.18。
微胶囊固化剂的制备
称取10g二氨基二苯基甲烷,逐渐加入到含有1000g甲苯的反应釜1中,加热到40℃,用框式桨搅拌50分钟,待固化剂完全溶解。称取40g聚合物a-2将其加入已配制好的固化剂溶液中,加热到40℃,用锚式搅拌桨搅拌45分钟。
另外的反应釜2中,加入1000g去离子水,并按加入水量的质量份数15g硬脂酸聚氧乙烯酯,在搅拌条件下使其完全溶解;在机械搅拌条件下,将反应釜1配制好的溶液逐滴滴加进反应釜2配制好的水溶液中;滴加完成后升高搅拌速度至1000r/min,保持9小时;待溶剂完全挥发后,停止搅拌。
将悬浊液,在6000r/min的速度下离心,得到下层浑浊液用无水乙醇超声洗涤20分钟,超声结束后,将烧杯中的固体物质用无水乙醇反复洗涤2-3次,过滤后,将固体置于50℃烘箱中8小时烘干,最后得到干燥的固体粉末,即微胶囊固化剂。
实施例3
微胶囊壁材料
聚合物b-1的制备
在装有搅拌器的三口瓶中,将183mg4-羟甲基-2硝基苄基醇(b1)溶解在90ml干燥二氯甲烷中,再加入1.83mg4-二甲基氨基吡啶。搅拌均匀后,加入4mg三乙胺后,滴加460mg2-溴-2-甲基-丙酰溴,室温搅拌,反应30小时,用18ml甲醇淬灭反应。向反应液中加入10ml四氢呋喃溶剂,用300目碱性硅胶过滤三乙胺盐酸盐,将滤液滴入乙醚中,析出沉淀,用布氏漏斗过滤,再多次用去离子水洗,110℃真空干燥13h得到396mg中间体b2,产率,83%。
质谱分析,[m+h]+:479.96;元素分析,c39.93h3.97br33.20n2.90o19.94。红外(cm-1):3004,2981,2934,1740,1533,1465,1363,1268,1154,1109。
在氮气气氛下向装有搅拌器的施兰克反应瓶中,加入481mg中间体b2和143mg溴化亚铜,加入10ml甲基丙烯酸甲酯,再滴加173mgn,n,n',n”,n”-五甲基二亚乙基三胺经过3次冷冻—抽真空—通氮气解冻后,在真空条件下封管,在油浴中将反应液加热到100℃,反应24小时。体系放凉后,将反应混合物倒入水中,得到聚合物,再用粉碎机粉碎成细颗粒,然后用布氏漏斗过滤,再多次用去离子水洗,乙醇洗后110℃真空干燥15h,得到产物聚合物b-1。
红外(cm-1):2977,2933,2879,1730,1539,1448,1394,1369,1258,1154,846,736。分子量:mn=40000,pdi=1.20。
微胶囊固化剂的制备
称取10g氨基二苯基砜逐渐加入到含有1000g二甲苯的反应釜1中,加热到40℃,用锚式桨搅拌50分钟,待固化剂完全溶解。称取40g聚合物b-1将其加入已配制好的固化剂溶液中,加热到40℃,用锚式搅拌桨搅拌45分钟。
另外的反应釜2中,加入1000g去离子水,并按加入水量的质量份数15g十二烷基硫酸钠,在搅拌条件下使其完全溶解;在机械搅拌条件下,将反应釜1配制好的溶液逐滴滴加进反应釜2配制好的水溶液中;滴加完成后升高搅拌速度至1000r/min,保持9小时;待溶剂完全挥发后,停止搅拌。
将悬浊液,在6000r/min的速度下离心,得到下层浑浊液用无水乙醇超声洗涤20分钟,超声结束后,将烧杯中的固体物质用无水乙醇反复洗涤2-3次,过滤后,将固体置于50℃烘箱中8小时烘干,最后得到干燥的固体粉末,即微胶囊固化剂。
实施例4
微胶囊壁材料
聚合物b-2的制备
在装有搅拌器的三口瓶中,将183mg4-羟甲基-2硝基苄基醇(b1)溶解在90ml干燥二氯甲烷中,再加入1.83mg4-二甲基氨基吡啶。搅拌均匀后,加入4mg三乙胺后,滴加460mg2-溴-2-甲基-丙酰溴,室温搅拌,反应30小时,用18ml甲醇淬灭反应。向反应液中加入10ml四氢呋喃溶剂,用300目碱性硅胶过滤三乙胺盐酸盐,将滤液滴入乙醚中,析出沉淀,用布氏漏斗过滤,再多次用去离子水洗,110℃真空干燥13h得到396mg中间体b2,产率,83%。
质谱分析,[m+h]+:479.96;元素分析,c39.93h3.97br33.20n2.90o19.94。红外(cm-1):3004,2981,2934,1740,1533,1465,1363,1268,1154,1109。
在氮气气氛下向装有搅拌器的施兰克反应瓶中,加入481mg中间体b2和143mg溴化亚铜,加入5ml甲基丙烯酸甲酯(pmma)和5ml苯乙烯(ps),再滴加173mgn,n,n',n”,n”-五甲基二亚乙基三胺经过3次冷冻—抽真空—通氮气解冻后,在真空条件下封管,在油浴中将反应液加热到100℃,反应24小时。体系放凉后,将反应混合物倒入水中,得到聚合物,再用粉碎机粉碎成细颗粒,然后用布氏漏斗过滤,再多次用去离子水洗,乙醇洗后110℃真空干燥15h,得到产物聚合物b-2。
红外(cm-1):3000,2933,2880,1741,1540,1445,1384,1369,1258,1154,846,736。分子量:mn=39800,pdi=1.25。
微胶囊固化剂的制备
称取10gn-氨乙基哌嗪,逐渐加入到含有1000g氯苯的反应釜1中,加热到40℃,用框式桨搅拌50分钟,待固化剂完全溶解。称取40g聚合物b-2将其加入已配制好的固化剂溶液中,加热到40℃,用锚式搅拌桨搅拌45分钟。
另外的反应釜2中,加入1000g去离子水,并按加入水量的质量份数15g硬脂酸聚氧乙烯酯,在搅拌条件下使其完全溶解;在机械搅拌条件下,将反应釜1配制好的溶液逐滴滴加进反应釜2配制好的水溶液中;滴加完成后升高搅拌速度至1000r/min,保持9小时;待溶剂完全挥发后,停止搅拌。
将悬浊液,在6000r/min的速度下离心,得到下层浑浊液用无水乙醇超声洗涤20分钟,超声结束后,将烧杯中的固体物质用无水乙醇反复洗涤2-3次,过滤后,将固体置于50℃烘箱中8小时烘干,最后得到干燥的固体粉末,即微胶囊固化剂。
实施例5
微胶囊壁材料
聚合物c-1的制备
在装有搅拌器的三口瓶中,将183mg5-羟甲基-2硝基苄基醇(c1)溶解在90ml干燥二氯甲烷中,再加入1.83mg4-二甲基氨基吡啶。搅拌均匀后,加入4mg三乙胺后,滴加463mg2-溴-2-甲基-丙酰溴,室温搅拌,反应24小时,用20ml甲醇淬灭反应。向反应液中加入10ml四氢呋喃溶剂,用500目碱性三氧化二铝过滤三乙胺盐酸盐,将滤液滴入乙醚中,析出沉淀,用布氏漏斗过滤,再多次用去离子水洗,100℃真空干燥15h得到394mg中间体c2,产率,82%。
质谱分析,[m+h]+:479.96;元素分析,c39.91h3.95br33.20n2.90o19.94。红外(cm-1):3004,2981,2934,1740,1533,1465,1362,1270,1154,1109。
在氮气气氛下向装有搅拌器的施兰克反应瓶中,加入481mg中间体c2和143mg溴化亚铜,加入10ml甲基丙烯酸甲酯,再滴加173mgn,n,n',n”,n”-五甲基二亚乙基三胺经过3次冷冻—抽真空—通氮气解冻后,在真空条件下封管,在油浴中将反应液加热到100℃,反应24小时。体系放凉后,将反应混合物倒入水中,得到聚合物,再用粉碎机粉碎成细颗粒,然后用布氏漏斗过滤,再多次用去离子水洗,乙醇洗后110℃真空干燥15h,得到产物聚合物c-1。
红外(cm-1):2977,2933,2880,1731,1539,1448,1394,1369,1258,1154,846,736。分子量:mn=40000,pdi=1.19。
微胶囊固化剂的制备
称取10g3-苯基-1,1-二甲基脲,逐渐加入到含有1000g氯仿的反应釜1中,加热到40℃,用框式桨搅拌50分钟,待固化剂完全溶解。称取40g聚合物c-1将其加入已配制好的固化剂溶液中,加热到40℃,用锚式搅拌桨搅拌45分钟。
另外的反应釜2中,加入1000g去离子水,并按加入水量的质量份数15g硬脂酸聚氧乙烯酯,在搅拌条件下使其完全溶解;在机械搅拌条件下,将反应釜1配制好的溶液逐滴滴加进反应釜2配制好的水溶液中;滴加完成后升高搅拌速度至1000r/min,保持9小时;待溶剂完全挥发后,停止搅拌。
将悬浊液,在6000r/min的速度下离心,得到下层浑浊液用无水乙醇超声洗涤20分钟,超声结束后,将烧杯中的固体物质用无水乙醇反复洗涤2-3次,过滤后,将固体置于50℃烘箱中8小时烘干,最后得到干燥的固体粉末,即微胶囊固化剂。
实施例6
微胶囊壁材料
聚合物c-2的制备
在装有搅拌器的三口瓶中,将183mg5-羟甲基-2硝基苄基醇(c1)溶解在90ml干燥二氯甲烷中,再加入1.83mg4-二甲基氨基吡啶。搅拌均匀后,加入4mg三乙胺后,滴加463mg2-溴-2-甲基-丙酰溴,室温搅拌,反应24小时,用20ml甲醇淬灭反应。向反应液中加入10ml四氢呋喃溶剂,用500目碱性三氧化二铝过滤三乙胺盐酸盐,将滤液滴入乙醚中,析出沉淀,用布氏漏斗过滤,再多次用去离子水洗,100℃真空干燥15h得到394mg中间体c2,产率,82%。
质谱分析,[m+h]+:479.96;元素分析,c39.91h3.95br33.20n2.90o19.94。红外(cm-1):3004,2981,2934,1740,1533,1465,1362,1270,1154,1109。
在氮气气氛下向装有搅拌器的施兰克反应瓶中,加入481mg中间体c2和143mg溴化亚铜,加入5ml甲基丙烯酸甲酯(pmma)和5ml苯乙烯(ps),再滴加173mgn,n,n',n”,n”-五甲基二亚乙基三胺经过3次冷冻—抽真空—通氮气解冻后,在真空条件下封管,在油浴中将反应液加热到100℃,反应24小时。体系放凉后,将反应混合物倒入水中,得到聚合物,再用粉碎机粉碎成细颗粒,然后用布氏漏斗过滤,再多次用去离子水洗,乙醇洗后110℃真空干燥15h,得到产物聚合物c-2。
红外(cm-1):2977,2933,2880,1731,1528,1447,1389,1370,1258,1154,846,736。分子量:mn=40100,pdi=1.19。
微胶囊固化剂的制备
称取10g己二胺,逐渐加入到含有1000g二甲苯的反应釜1中,加热到40℃,用锚式桨搅拌50分钟,待固化剂完全溶解。称取40g聚合物c-2将其加入已配制好的固化剂溶液中,加热到40℃,用锚式搅拌桨搅拌45分钟。
另外的反应釜2中,加入1000g去离子水,并按加入水量的质量份数15g十二烷基硫酸钠,在搅拌条件下使其完全溶解;在机械搅拌条件下,将反应釜1配制好的溶液逐滴滴加进反应釜2配制好的水溶液中;滴加完成后升高搅拌速度至1000r/min,保持9小时;待溶剂完全挥发后,停止搅拌。
将悬浊液,在6000r/min的速度下离心,得到下层浑浊液用无水乙醇超声洗涤20分钟,超声结束后,将烧杯中的固体物质用无水乙醇反复洗涤2-3次,过滤后,将固体置于50℃烘箱中8小时烘干,最后得到干燥的固体粉末,即微胶囊固化剂。
实施例7
以实施例6的微胶囊壁材料为聚合物c-2和聚甲基丙烯酸甲酯(mn,8000-10000)。
微胶囊固化剂的制备
称取10g己二胺,逐渐加入到含有1000g二甲苯的反应釜1中,加热到40℃,用锚式桨搅拌50分钟,待固化剂完全溶解。称取20g聚合物c-2和10g聚甲基丙烯酸甲酯(mn,8000-10000)将它们加入已配制好的固化剂溶液中,加热到40℃,用锚式搅拌桨搅拌45分钟。
另外的反应釜2中,加入1000g去离子水,并按加入水量的质量份数15g十二烷基硫酸钠,在搅拌条件下使其完全溶解;在机械搅拌条件下,将反应釜1配制好的溶液逐滴滴加进反应釜2配制好的水溶液中;滴加完成后升高搅拌速度至1000r/min,保持9小时;待溶剂完全挥发后,停止搅拌。
将悬浊液,在6000r/min的速度下离心,得到下层浑浊液用无水乙醇超声洗涤20分钟,超声结束后,将烧杯中的固体物质用无水乙醇反复洗涤2-3次,过滤后,将固体置于50℃烘箱中8小时烘干,最后得到干燥的固体粉末,即微胶囊固化剂。
对上述实施例制备的微胶囊固化剂进行检测:
1、扫描电子显微镜
对各实施例1-6的旋转蒸发后的微胶囊粉末进行喷金处理,用扫描电子显微镜(sem)进行观察拍照。结果如图2-7所示。微胶囊粉末的粒径尺寸大约在5-30μm之间。
2、微胶囊固化剂释放性能的测定
以体积比为1:1的乙醇水溶液作为缓释介质,采用透析袋法测定微胶囊的释放性能。称取25mg实施例6中得到的微胶囊固化剂置于盛有3ml缓释介质的透析袋中,将袋口密封后置于装有400ml缓释介质的锥形瓶中,紫外光照射下分别每隔一段时间取缓释介质1ml用高效液相色谱测定样品中己二胺含量,并立即补充1ml缓释介质,最后绘制其累积释放曲线,结果如图8所示,在紫外光照射下,该微胶囊可室温在3min内100%释放己二胺固化剂。
显然,上述实施例仅仅是为清楚地说明所作的举例,而并非对实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。而由此所引伸出的显而易见的变化或变动仍处于本发明创造的保护范围之中。
- 还没有人留言评论。精彩留言会获得点赞!