C4′-氟代尿苷亚磷酰胺单体和C4′-氟代尿苷修饰RNA的制备方法与流程
本发明涉及寡聚核苷酸的化学合成领域,具体涉及c4′-氟代尿苷亚磷酰胺单体以及c4′-氟代尿苷定点修饰rna的制备方法。
背景技术:
氟代修饰的rna在生物医学领域有广泛的应用前景[org.biomol.chem.,2017,15,9552–9565]。c2′-氟代修饰的rna成功应用于核酸适配体、小干扰rna等,可以增强稳定性,提高效力[nature,2011,478,404-408]。此外,c4′-氟修饰核苷类似物nucleocidin是天然存在的五种含氟天然产物之一,具有良好的抗代谢活性,c4′-氟代修饰的核苷也因此引起了关注。guillerm[bioorg.med.chem.lett.1995,5,1455-1460]和verdine[org.lett.,2007,9,5007-5009]分别发展了各自的方法合成了c4′-氟代核苷。然而c4′-氟代核苷不稳定,将c4′-氟代核苷转化为c4′-氟亚磷酰胺单体并制备c4′-氟代修饰的rna一直没有成功,限制了对其生化性质的进一步探讨。
技术实现要素:
本发明的目的是针对c4′-氟代核苷不稳定的实际问题,提供一种通过分阶段、选择性保护核糖核苷中羟基的策略,即c4′-氟代尿苷亚磷酰胺单体及其制备方法,以及利用c4′-氟代尿苷磷酰胺单体制备c4′-氟代尿苷定点修饰的rna的方法。
本发明技术方案
1、为了实现上述目的,本发明第一方面提供了一种c4′-氟代尿苷亚磷酰胺单体,其中,该c4′-氟代尿苷亚磷酰胺单体的化学结构式为:
式(1)所示的化合物可以称为2′-o-(叔丁基二甲基硅烷基)-3′-[(2-氰基乙氧基)-n,n′-二异丙基亚磷酰胺]-4′-c-氟-5′-o-二甲氧基三苯甲基尿苷。
其中,在式(1)中,“dmtr-”为二甲氧基三苯甲基,“n(ipr)2-”为n,n-二异丙基胺基,“-tbdms”为叔丁基二甲基硅烷基。
2、本发明第二方面提供了一种c4′-氟代尿苷亚磷酰胺单体的制备方法,该方法包括以下步骤:
(1)将4′-c-氟-5′-碘-5′-脱氧尿苷与硝酸银、吡啶和四氢呋喃形成第一混合溶液;之后将所述第一混合溶液与叔丁基二甲基氯硅烷进行反应,将反应后得到的溶液旋蒸除去溶剂,用二氯甲烷溶解后洗涤、过滤、干燥、柱层析得到2′-o-(叔丁基二甲基硅烷基)-4′-c-氟-5′-碘-5′-脱氧尿苷。
(2)将步骤(1)得到的2′-o-(叔丁基二甲基硅烷基)-4′-c-氟-5′-碘-5′-脱氧尿苷与4-二甲氨基吡啶、吡啶进行第二混合,形成第二混合溶液;再将所述第二混合溶液与乙酸酐进行反应;旋蒸除去溶剂,用二氯甲烷溶解后洗涤、过滤、干燥、柱层析得到2′-o-(叔丁基二甲基硅烷基)-3′-乙酸酯-4′-c-氟-5′-碘-5′-脱氧尿苷。
(3)将三氟乙酸与四正丁基氢氧化铵溶液混合调节ph后,加入步骤(2)得到的2′-o-(叔丁基二甲基硅烷基)-3′-乙酸酯-4′-c-氟-5′-碘-5′-脱氧尿苷、二氯甲烷进行第三混合,形成第三混合溶液;再将所述第三混合溶液与间氯过氧苯甲酸进行反应;然后将反应得到的有机相洗涤、过滤、干燥、柱层析得到2′-o-(叔丁基二甲基硅烷基)-3′-乙酸酯-4′-c-氟代尿苷。
(4)将步骤(3)得到的2′-o-(叔丁基二甲基硅烷基)-3′-乙酸酯-4′-c-氟代尿苷与氨/甲醇溶液进行反应;旋蒸除去溶剂,柱层析得到产物。
(5)将步骤(4)得到的产物与4-二甲氨基吡啶、吡啶以及4,4′-二甲氧基三苯甲基氯进行反应;旋蒸除去溶剂,用二氯甲烷溶解后然后将有机相洗涤、过滤、干燥、柱层析得到2′-o-(叔丁基二甲基硅烷基)-4′-c-氟-5′-o-二甲氧基三苯甲基尿苷。
(6)将步骤(5)得到的2′-o-(叔丁基二甲基硅烷基)-4′-c-氟-5′-o-二甲氧基三苯甲基尿苷与二氯甲烷、n,n′-二异丙基乙胺以及2-甲基咪唑进行第四混合,形成第四混合溶液;再将所述第四混合溶液与(2-氰基乙氧基)-n,n′-二异丙基亚磷酰氯进行反应;旋蒸除去溶剂,用二氯甲烷溶解后然后将有机相洗涤、过滤、干燥、柱层析得到2′-o-(叔丁基二甲基硅烷基)-3′-[(2-氰基乙氧基)-n,n′-二异丙基亚磷酰胺]-4′-c-氟-5′-o-二甲氧基三苯甲基尿苷,即本发明所述的c4′-氟代尿苷亚磷酰胺单体。
优选地,在步骤(1)中,所述4′-c-氟-5′-碘-5′-脱氧尿苷与所述硝酸银、所述吡啶、所述四氢呋喃、所述叔丁基二甲基氯硅烷用量的比为1mmol:3.5mmol:4ml:4ml:3.5mmol;以及,
所述反应的条件包括:温度为18-25℃,时间为1.5-2小时,搅拌速度为250-500转/分钟。
优选地,在步骤(2)中,所述2′-o-(叔丁基二甲基硅烷基)-4′-c-氟-5′-碘-5′-脱氧尿苷与所述4-二甲氨基吡啶、所述吡啶、所述乙酸酐用量的比为1mmol:0.3mmol:10ml:5mmol;以及,
所述乙酸酐以滴加的方式加入所述第二混合溶液中,所述乙酸酐的滴加速率为15滴/分钟;以及,
所述反应的条件包括:温度为18-25℃,时间为0.5-1小时,搅拌速度为250-500转/分钟。
优选地,在步骤(3)中,所述三氟乙酸与所述四正丁基氢氧化铵溶液、所述2′-o-(叔丁基二甲基硅烷基)-3′-乙酸酯-4′-c-氟-5′-碘-5′-脱氧尿苷、所述二氯甲烷、所述间氯过氧苯甲酸的用量比为0.8ml:10ml:1mmol:10ml:5mmol;其中所述四正丁基氢氧化铵溶液的质量浓度为55%,所述间氯过氧苯甲酸的纯度为85%;
所述三氟乙酸以滴加的方式加入所述四正丁基氢氧化铵溶液中调ph为4;所述间氯过氧苯甲酸分三次加入第三混合溶液中,间隔时间为0.5小时;以及,
所述反应的条件包括:温度为18-25℃,时间为40-46小时,搅拌速度为250-500转/分钟。
优选地,在步骤(4)中,所述2′-o-(叔丁基二甲基硅烷基)-3′-乙酸酯-4′-c-氟代尿苷与氨/甲醇溶液用量比为1mmol:8ml,其中氨/甲醇溶液的物质的量浓度为7mol/l;以及,
所述反应的条件包括:温度为18-25℃,时间为3-4小时,搅拌速度为250-500转/分钟。
优选地,在步骤(5)中,所述产物与所述4-二甲氨基吡啶、所述吡啶以及4,4′-二甲氧基三苯甲基氯的用量比为1mmol:0.3mmol:8ml:1.3mmol;以及,
所述反应的条件包括:温度为40-50℃,时间为3-5小时,搅拌速度为250-500转/分钟。
优选地,在步骤(6)中,所述2′-o-(叔丁基二甲基硅烷基)-4′-c-氟-5′-o-二甲氧基三苯甲基尿苷与所述二氯甲烷、所述n,n′-二异丙基乙胺、所述2-甲基咪唑、所述(2-氰基乙氧基)-n,n′-二异丙基亚磷酰氯的用量比为1mmol:20ml:10mmol:4mmol:5mmol;以及,
所述反应的条件包括:温度为18-25℃,时间为15分钟,搅拌速度为250-500转/分钟。
3、本发明第三方面提供了一种c4′-氟代尿苷定点修饰rna的制备方法,其中,该制备方法中的合成试剂包括以上所述的制备方法制备得到的c4′-氟代尿苷亚磷酰胺单体。
优选地,该制备方法在abi394dna/rna合成仪上进行以得到寡聚核苷酸,其中:
优选地,所述合成试剂还包括叔丁基二甲基氯硅烷保护2′-oh的ac-rc、bz-ra、ac-rg和ru;
优选地,三氯乙酸/二氯甲烷用于脱保护;
优选地,capa试剂为乙酸酐、吡啶和四氢呋喃的组合物,且所述乙酸酐、所述吡啶和所述四氢呋喃的体积比为10体积%:(5-15)体积%:(75-85)体积%;
优选地,capb试剂为n-甲基咪唑和四氢呋喃的组合物,且所述n-甲基咪唑和所述四氢呋喃的体积比为10体积%:90体积%;
优选地,活化剂为乙硫基四唑和乙腈的组合物;
优选地,氧化剂为碘、水、吡啶和四氢呋喃的组合物,且所述碘、所述水、所述吡啶和所述四氢呋喃的用量比为0.02m:(1-2)体积%:(10-30)体积%:(75-85)体积%;以及所述,
优选地,所述c4’-氟代rna亚磷酰胺单体的偶合时间为0.5-1小时;
优选地,将上述固相合成得到的寡聚核苷酸用0.75ml浓度为28重量%的氨水混合0.25ml无水乙醇在55℃下处理10-12h,然后,取出上清液离心浓缩后,加入0.5ml三乙胺三氢氟酸盐处理20-24h,经c18柱脱盐处理后,离心浓缩,采用20重量%的变性聚丙烯凝胶电泳纯化。
本发明的优点和有益效果是:(1)通过分阶段、选择性保护尿苷中羟基的策略,成功制备了c4′-氟代尿苷亚磷酰胺单体;(2)c4′-氟代尿苷亚磷酰胺单体成功应用于制备c4′-氟代尿苷定点修饰的rna。本发明所述的c4′-氟代尿苷定点修饰的rna的制备方法,在研究c4′-氟代rna的生化性质等方面有着广泛的应用前景。
附图说明:
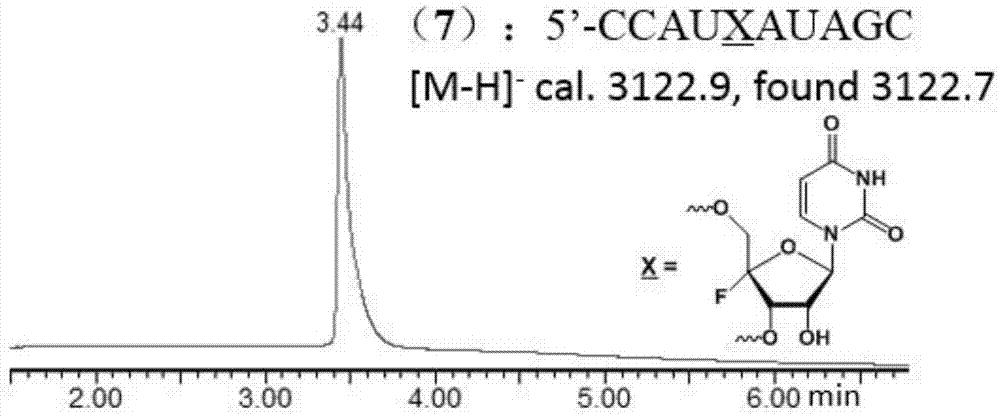
图1为uplc-ms分析c4′-氟代尿嘧啶核苷修饰rna分子(7)的纯度。
具体实施方式
以下对本发明的具体实施方式进行详细说明。此处所描述的具体实施方式仅用于说明和解释本发明,并不用于限制本发明。
4′-c-氟-5′-碘-5′-脱氧尿苷(自制);二氯甲烷(天津市河东区广达服务部,货号1220);na2co3·10h2o晶体(天津市河东区广达服务部,货号2093);氯化钠(天津市化学试剂供销公司,货号017);乙酸酐(天津市化学试剂供销公司,货号282);吡啶[阿拉丁试剂(上海)有限公司,货号p111511];四氢呋喃(北京华威锐科化工有限公司,货号hwmt818767);甲醇(北京百灵威科技有限公司,货号980290-500ml)、55%(w/w)四正丁基氢氧化铵水溶液(北京百灵威科技有限公司,货号16198-50g);氨水/甲醇(2n)溶液(alfa,货号h27080);氨水/甲醇(7n)溶液(alfa公司,货号h30382);2-甲基咪唑[梯希爱(上海)化成工业发展有限公司,货号m0508]、n,n-二异丙基乙胺[梯希爱(上海)化成工业发展有限公司,货号d1599];叔丁基二甲基氯硅烷[梯希爱(上海)化成工业发展有限公司,货号b0995];硝酸银(安耐吉,货号a01w5100190250)、间氯过氧苯甲酸(安耐吉,货号a01w520002-100g)、2-氰乙基n,n-二异丙基氯亚磷酰胺(安耐吉,货号a07m901005-01);4-二甲氨基吡啶(adamas,货号14766b)、4,4”-双甲氧基三苯甲基氯(adamas,货号55311c);三氟乙酸(acros,货号293810250)等原料为市售品。
实施例1
c4′-氟代尿嘧啶亚磷酰胺单体的合成。
其合成步骤如下:
步骤一、化合物(3)的制备:在氩气保护下,将式(2)对应单体(371.9mg,1mmol,1.0eq.)溶于4ml干燥的四氢呋喃,加入4ml干燥的吡啶和527.5mg(3.5mmol,3.5eq.)叔丁基二甲基氯硅烷,594.5mg(3.5mmol,3.5eq.)硝酸银。室温下搅拌1.5小时。旋干溶剂,加入20ml二氯甲烷稀释,依次用20ml饱和碳酸氢钠水溶液、饱和氯化钠水溶液清洗,有机层用无水硫酸镁干燥,用乙酸乙酯/石油醚(梯度,v/v,1:4~1:2~1:1)柱层析,得白色固体式(3)对应单体338.5mg(0.7mmol,两步产率70重量%)。
1hnmr(400mhz,dmso-d6)δ(ppm)11.52(s,1h,nh),7.69(d,j=7.87hz,1h,h-6,=ch),5.95(s,1h,h-1’),5.71(d,j=7.55hz,1h,h-5),5.34(d,j=8.52hz,1h,3’-oh),4.42(m,h-3’),4.34(m,h-2’),3.56(m,2h,h-5’,h-5”),0.84(s,9h,tbu),0.05(s,3h,-ch3),0.04(s,3h,-ch3);13cnmr(100.6mhz,cdcl3)δ(ppm)163.23,149.52,142.57,114.18(d,jh-f=229.69hz,1c,c-4’),103.31,97.49,73.22,71.22(d,jc-f=23.24hz,1c,c-3’),25.56,17.99,2.29(d,jc-f=33.48hz,1c,c-5’),-4.87,-5.22;19fnmr(376mhz,dmso-d6)δ(ppm)-110.27.m/z:calcdforc15h24fin2o5si[m+h]+,487.0561;found,487.0572.
步骤二、化合物(4)的制备:称取48.5mg(0.1mmol,1.0eq.)式(3)所示的单体溶于1ml干燥的吡啶。在氩气保护下,加入4mg(0.03mmol,0.3eq.)4-二甲氨基吡啶。在冰浴条件(0℃)及搅拌下,将0.05ml(0.5mmol,5.0eq.)乙酸酐缓慢滴加至反应体系,升至室温搅拌1小时。旋干溶剂,加入5ml二氯甲烷稀释,依次用5ml饱和碳酸氢钠水溶液、饱和氯化钠水溶液清洗,有机层用无水硫酸镁干燥,旋干溶剂,用乙酸乙酯/石油醚(梯度,v/v,1:8~1:4~1:2)柱层析,得白色固体式(4)对应单体52.1mg(0.098mmol,产率98重量%)。
1hnmr(400mhz,dmso-d6)δ(ppm)9.87(s,1h,nh),7.30(d,j=7.96hz,1h,h-6,=ch),5.82(m,2h,h-1’andh-5),5.39(d,j=6.70hz,jh-f=14.48hz,1h,h-3’),4.67(m,h-2’),3.58(dd,1h,j=7.95hz),jh-f=11.16hz,h-5”),2.17(s,3h,-ch3),0.86(s,9h,tbu),0.01(s,3h,-ch3),0.003(s,3h,-ch3);13cnmr(100.6mhz,dmso-d6)δ(ppm)169.8,163.1,149.8,141.3,113.6(d,1c,jc-f=237.38hz,c-4’),103.5,94.67,72.3,71.6(d,jc-f=19.59hz,1c,c-3’),25.36,20.53,17.78,3.26(d,jc-f=35.30hz,1c,c-5’),-4.99,-5.31.m/z:calcdforc17h26fin2o6si[m+na]+,529.0667;found,529.0639.
步骤三、化合物(5)的制备:在反应容器中,滴加三氟乙酸到496μl(1.86mmol,20eq.)四正丁基氢氧化铵(体积浓度55%)中调ph至4,将49.3mg(0.093mmol,1.0eq.)式(4)对应的单体溶于2ml二氯甲烷,加入至反应体系,分三次加入80mg(0.46mmol,5.0eq.)间氯过氧苯甲酸(间隔30min),常温搅拌46h。加入2ml饱和硫代硫酸钠水溶液淬灭反应,依次用5ml饱和碳酸氢钠水溶液溶液、5ml饱和氯化钠水溶液清洗,用丙酮/乙酸乙酯/石油醚(v/v/v,1:1:5)柱层析,得白色固体式(5)所对应的单体29.5mg(0.070mmol,产率75.8重量%)。
1hnmr(400mhz,dmso)δ(ppm)11.54(s,1h,-nh),7.80(d,j=8.12hz,1h,h-6,=ch),6.01(d,j=4.33hz1h,h-1’),5.75(m,1h,h-5),5.33(dd,j=5.33hz,jh-f=13.37hz1h,h-3’),4.58(d,j=4.41hz,j=6.23hz,1h,h-2’),3.61(m,2h,h-5’,h-5”),2.11(s,3h,ome),0.81(s,9h,tbu),-0.02(s,3h,-ch3),-0.01(s,3h,-ch3);13cnmr(100.6mhz,dmso-d6)δ(ppm)169.72,163.33,150.78,141.15,116.40(d,1c,jh-f=235.53hz,c-4’),103.18,91.33,71.79,70.27(d,jc-f=17.37hz,1c,c-3’),61.33(d,jc-f=39.38hz,1c,c-5’),25.77,20.88,17.99,-4.85,-5.05.m/z:calcdforc17h27fin2o7si[m+h]+,419.1650;found,419.1641.
步骤四、化合物(6)的制备:在氩气保护下,将144.3mg(0.34mmol,1.0eq.)式(5)所对应的单体加入到3ml(7n)的氨/甲醇中,常温搅拌3h。旋干除去溶剂,用乙酸乙酯/石油醚(梯度,v/v,1:4~1:2~1:1)柱层析,将得到rf=0.34(丙酮/乙酸乙酯/石油醚=1:1:4)的组分溶于3ml无水吡啶中,加入11mg(0.09mmol,0.3eq.)4-二甲氨基吡啶、132.1mg(0.4mmol,1.3eq.)二甲氧基三苯甲基,40℃搅拌4小时,1ml甲醇淬灭反应,旋干溶剂,二氯甲烷溶解后饱和碳酸氢钠水溶液清洗,用乙酸乙酯/石油醚(梯度,v/v,1:8~1:4~1:2~1:1,加入0.5体积%的三乙胺)柱层析,得式(6)对应的单体140.5mg(0.204mmol,产率68重量%)。
1hnmr(400mhz,dmso-d6)δ(ppm)11.56(s,1h,-nh),7.82(d,j=8.06hz,1h,h-6,=ch),7.41-7.26(m,9h,dmtr-arh),6.92(s,2h,dmtr-arh),6.90(s,2h,dmtr-arh),6.00(s,1h,h-1’),5.46(d,j=8.01hz,1h,h-5),5.14(d,j=9.07hz,1h,-oh),4.61(m,1h,h-3’),4.56(d,j=6.08hz,1h,h-2’),3.74(s,6h,-ome),3.30(m,1h,h-5’),3.22(m,1h,h-5”),0.88(s,9h,tbu),0.11(s,3h,-ch3),0.10(s,3h,-ch3);13cnmr(100.6mhz,dmso-d6)δ(ppm)162.94,158.21,150.06,144.23,141.29,134.80,134.68,129.75,127.89,127.63,126.84,116.67(d,jh-f=229.69hz,1c,c-4’),113.24,102.05,92.79,86.10,72.92,69.40(d,jc-f=20.57hz,1c,c-3’),63.42,54.97,29.98,25.55,-4.87,-5.22。m/z:calcdforc36h43fn2nao8si[m+na]+,701.2670;found,701.2670.
步骤五、化合物(1)的制备:称取2.16g(3.16mmol,1.0eq.)式(6)所对应的单体,在氩气保护下溶于60ml二氯甲烷中。依次加入4.5ml(31.6mmol,10eq.)n,n’-二异丙基乙胺,1ml(12.64mmol,4eq.)2-甲基咪唑,2.4ml(15.8mmol,5eq.)(2-氰基乙氧基)-n,n’-二异丙基亚磷酰氯,常温下搅拌15分钟。反应结束后,在氩气保护下将体系减压蒸干,然后用少量二氯甲烷溶解,快速柱层析丙酮/乙酸乙酯/石油醚(v/v/v,1:1:5,+0.5体积%三乙胺)纯化得到浅黄色固体式(1)所对应的单体2.686g(3mmol,产率95重量%)。产物有2个异构体,比例为:2.25:1。
1hnmr(400mhz,cdcl3)δ(ppm)7.81(d,j=8.07hz,0.64h,h-6,=ch),7.66(d,j=8.12hz,0.26h,h-6,=ch)7.26-7.10(m,9h,dmtr-arh),6.72(s,2h,dmtr-arh),6.70(s,2h,dmtr-arh),6.09(s,0.26h,h-1’),5.96(s,0.61h,h-1’)5.07(d,j=8.01hz,1h,h-5),4.40(m,1h,h-3’),4.28(m,0.28h,h-2’),4.19(d,j=4.80hz0.63h,h-2’),3.66(s,6h,-ome),3.46(m,4h),2.52(m,0.68h),2.37(m,1.3h),1.02(m,12h,4ch3),0.89(d,j=6.60hz,2h)0.93(s,9h,tbu),0.05-0.00(m,6h,-ch3);31pnmr(162mhz,cd3cl)δ:152.2(s,0.27p),149.6(s,0.64p)。m/z:calcdforc45h61fn4o9psi[m+h]+,879.3929;found,879.3942.
实施例2
本实施例在于说明由实施1制备的c4′-氟代尿嘧啶核苷亚磷酰胺单体(1)进行rna固相合成rna序列(7):5′-ccauxauagc。在序列(7)中,x为c4′-氟代尿嘧啶核苷。
在abi394dna/rna合成仪上通过常规程序和试剂进行1μmol寡聚核苷酸合成。合成试剂包括ac-rc,bz-ra,ac-rg,ru和实施例1制备的c4’-氟代尿嘧啶亚磷酰胺单体(1);3重量%三氯乙酸/二氯甲烷用于脱保护;capa试剂:10体积%乙酸酐/10体积%吡啶/80体积%四氢呋喃(v/v/v),capb试剂:10体积%n-甲基咪唑/90体积%四氢呋喃(v/v);活化剂:0.25m乙硫基四唑/乙腈;氧化剂:0.02m碘/2体积%水/20体积%吡啶/78体积%四氢呋喃(v/v/v)。c4’-氟代尿嘧啶核苷亚磷酰胺单体的偶合时间是1小时。
将上述固相合成得到的rna分子用0.75ml(28重量%)氨水混合0.25ml无水乙醇在55℃下处理12h,然后,取出上清液离心浓缩后,加入0.5ml三乙胺三氢氟酸盐处理24h,经c18柱脱盐处理后,离心浓缩,采用20重量%的变性聚丙烯凝胶电泳纯化。得到纯的rna(7)经过了uplc-ms分析鉴定(图1)。
可见,实施例1制备的c4’-氟代尿嘧啶亚磷酰胺单体(1)可以用于常规的rna固相合成与纯化。
以上详细描述了本发明的优选实施方式,但是,本发明并不限于此。在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,包括各个技术特征以任何其它的合适方式进行组合,这些简单变型和组合同样应当视为本发明所公开的内容,均属于本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!